甘草次酸固体脂质纳米凝胶的制备及体外透皮效应
2014-05-26宋艳丽韩腾飞李莎莎危红华郝保华
宋艳丽, 徐 坤, 韩腾飞, 李莎莎, 危红华, 程 亮, 郝保华
(西北大学生命科学学院,陕西西安 710069)
甘草次酸固体脂质纳米凝胶的制备及体外透皮效应
宋艳丽, 徐 坤, 韩腾飞, 李莎莎, 危红华, 程 亮, 郝保华*
(西北大学生命科学学院,陕西西安 710069)
目的 制备甘草次酸固体脂质纳米凝胶并考察其体外透皮效应。方法 采用微乳液法制备甘草次酸固体脂质纳米粒并考察其包封率、粒径与表面电位,以研和法制备固体脂质纳米粒凝胶;采用改良Franz立式扩散池法进行体外透皮实验,HPLC法测定甘草次酸,评价甘草次酸固体脂质纳米粒凝胶的经皮渗透结果。结果 甘草次酸固体脂质纳米粒外观为圆球形或椭球形;甘草次酸固体脂质纳米粒的包封率为 (64.75±1.36)%,粒径范围(46.13±20.10)nm,电位分布范围为 (-53.4±7.11)mV。24 h甘草次酸固体脂质纳米粒凝胶较甘草次酸固体脂质纳米粒的累积透过量提高66%。结论 甘草次酸固体脂质纳米粒凝胶能提高甘草次酸的透皮速率,有望成为甘草次酸透皮给药的新型制剂。
甘草次酸;固体脂质纳米粒;凝胶;体外透皮
甘草次酸 (glycyrrhetinic acid,GA)是豆科植物甘草的主要活性成分[1],主要从甘草的根部或根茎提取而得,属五环三萜皂苷类化合物[2]。甘草次酸具有明显的抗炎抗菌、抗肿瘤、抗病毒、抗氧化与免疫调节等多方面的药理活性[1],临床常用于治疗湿疹、荨麻疹、皮炎等皮肤病,但由于其为强的亲脂性成分,制成普通制剂经皮给药其渗透率低[3]。凝胶经皮给药制剂可以避免肝脏的首关效应,从而提高药物的生物利用度[4]。故本实验将甘草次酸制备为经皮给药制剂固体脂质纳米粒凝胶。
固体脂质纳米粒[5](solid lipid nanoparticles,SLNs)是采用可生物降解的载体材料制成的具有良好的缓控释作用和靶向性的一种剂型;生产过程不添加有机溶剂,克服了产品中有机溶剂不能完全挥尽的缺点。固体脂质纳米粒给药系统主要用于水不溶性药物[6]或者毒性大[7]的药物,其中多数为抗肿瘤药物[8-9]。
凝胶剂具有良好的生物相容性,对药物释放具有缓释、控释作用,且制备工艺简单而外形美观[10],文献报道凝胶作为给药基质,有促进透皮速率和在皮肤中的滞留量的作用[11-13]。刘丽宏[14]等以凝胶为载体制备了氯麻鼻用凝胶,该制剂提高了药物的生物利用度且延长了药物的作用时间。因此,本实验将甘草次酸制备为固体脂质纳米粒凝胶剂,以其提高药物的经皮透过量,为甘草次酸新型制剂的开发研究提供实验依据。
1 仪器、试药与动物
1.1 仪器 78-1型磁力加热搅拌器 (江苏省金坛市正基仪器有限公司);AG-285电子天平 (德国METTLER公司);AH-系列高压均质机 (ATSEn-gineering Inc.);ZS90纳米粒度及Zeta电位分析仪(英国Malvern公司);岛津LC-20AT高效液相色谱仪 (日本岛津公司);JEM-2000EX透射电子显微镜(日本电子公司)。
1.2 药品和试剂 甘草次酸 (质量分数≥98%,西安小草植物科技有限责任公司);大豆卵磷脂(天津市博迪化工有限公司);单硬脂酸甘油酯(上海山浦化工有限公司);吐温-80(成都市科隆化工试剂厂);卡波姆934P(美国誉誊公司);甲醇(Sigma,色谱纯);其他试剂均为分析纯;0.22 μm的微孔滤膜 (上海新亚药业有限公司)。
1.3 动物 小鼠,雄性,清洁级 (合格证号:陕西动证2006105),购自西安交通大学医学院。
2 甘草次酸固体脂质纳米粒凝胶的制备
2.1 甘草次酸固体脂质纳米粒的制备
2.1.1 空白固体脂质体纳米粒的制备 采用微乳液法制备空白固体脂质纳米粒,将处方量0.6 mL乳化剂吐温-80置于烧杯中,加46.2 mL蒸馏水超声分散至完全溶解,置于恒温水浴中,得到水相。将处方量的脂质单硬脂酸甘油酯2.0 g与大豆磷脂1.0 g在恒温磁力搅拌器上加热融化,在磁力搅拌器搅拌下,水相以10 mL/min的体积流量加到油相中,继续搅拌30 min,停止加热,得初分散体系。将初分散体系在AH-系列高压均质机上60 MPa乳匀6次,过0.22 μm滤膜,即得。
2.1.2 甘草次酸固体脂质纳米粒的制备 采用微乳液法制备甘草次酸固体脂质纳米粒,将处方量0.6 mL乳化剂吐温-80置于烧杯中,加46.2 mL蒸馏水超声分散至完全溶解,置于恒温水浴中,得到水相。将处方量甘草次酸0.1 g与脂质单硬脂酸甘油酯2.0 g与大豆磷脂1.0 g在恒温磁力搅拌器上加热融化,在磁力搅拌器搅拌下,水相以10 mL/min的体积流量加到油相中,继续搅拌30 min,停止加热,得初分散体系。将初分散体系在AH-系列高压均质机上60 MPa乳匀6次,过0.22 μm滤膜,即得甘草次酸固体脂质纳米粒。
2.2 甘草次酸固体脂质纳米粒理化考察
2.2.1 GA-SLNs外观形态 取甘草次酸固体脂质纳米粒浮悬液适量,用蒸馏水稀释至一定倍数后,滴至专用铜网上,用20 g/L磷钨酸复染,于透射电子显微镜下观察样品形态,结果见图1。
甘草次酸–固体脂质纳米粒透射电镜照片显示甘草次酸固体脂质纳米粒呈球形或椭球形,粒子分布比较均匀且无相互黏连。

图1 甘草次酸固体脂质纳米粒的透射电镜照片Fig.1 TEM of GA-SLNs
2.2.2 GA-SLNs粒径分布与表面电位 取甘草次酸-固体脂质纳米粒混悬液1 mL用双蒸水稀释至30 mL,置于马尔文ZS90纳米粒度及Zeta电位分析仪中,测量温度25℃,分散介质水的折光率为1.333,黏度为0.887 2 cP,激光入射角度90°,波长635 nm,进行粒度测定,用Zeta电位测定专用比色皿进行Zeta电位测定,每个样品进行3次测定,取平均值。粒径分布见图2。Zata电位分布见图3。
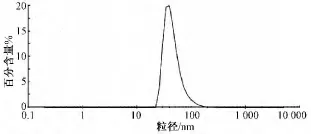
图2 甘草次酸-固体脂质纳米粒的粒径分布Fig.2 Particle size distribution of GA-SLNs
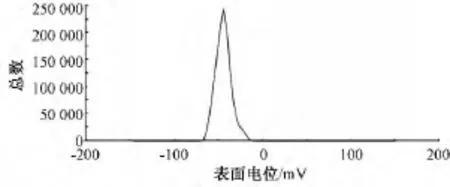
图3 甘草次酸-固体脂质纳米粒的表面电位图Fig.3 Particle surface potential of GA-SLNs
由图2可知平均粒径在57.19 nm,粒径范围(46.13±20.10)nm,粒径分布较窄。
由图3可知甘草次酸-固体脂质纳米粒表面带有负电位,大多数集中在-53.4 mV,电位分布范围为 (-53.4±7.11)mV。当表面电荷的绝对值大于30 mV时,代表分散体系比较稳定。2.2.3 包封率的测定 精密量取1 mL甘草次酸-固体脂质纳米粒,上Sephadex-G50柱 (250 mm×16 mm),以水为洗脱液,加甲醇超声破乳,定容,静置,取上清液进样。上清液经0.22 μm微孔滤膜过滤后,取续滤液采用“3.1”项下色谱条件测定,测得的峰面积带入标准曲线计算药物浓度。
包封率的计算公式为:EE%=C纳米粒/C总药量×100%。所制得甘草次酸固体脂质纳米粒的平均包封率为64.75%。
2.3 甘草次酸固体脂质纳米粒凝胶的制备
2.3.1 空白凝胶剂的制备 称取0.2 g卡波姆,加水10 mL溶胀24 h,加入0.8 mL乙醇后再继续滴加三乙醇胺至中性成凝胶,边滴加边搅拌,最后加水至20 mL,即得。前期做实验时制备相同比例的卡波姆与乙醇多份,开始时均采用pH试纸测定,在pH大约为6时,记录采用三乙醇胺的滴数,第一份加入一滴后,取出其中0.5 g,加入50 mL水,加热至80℃,搅匀,放冷,用pH计测定,记录 pH;第二份加入两滴后,取出其中0.5 g,加入50 mL水,加热至80℃,搅匀,放冷,用pH计测定,记录pH;依此类推直到pH 7为止。采用此法进行pH控制。
2.3.2 甘草次酸凝胶剂的制备 称取0.2 g卡波姆,加水10 mL溶胀24 h。称取甘草次酸0.02 g,溶解在0.8 mL的乙醇中,药物的乙醇溶液倒入溶胀后的卡波姆,滴加三乙醇胺至中性成凝胶,边滴加边搅拌,最后加水至20 mL,即得。
2.3.3 纳米粒凝胶的制备 按照“2.3.1”项制备空白凝胶,与“2.1.2”项制备固体脂质纳米粒1∶1混合,即得含药0.1%的纳米粒凝胶。
2.4 甘草次酸固体脂质纳米粒质量控制
2.4.1 纳米粒凝胶的性状 本品为乳白色透明凝胶,稠度适宜,涂展性好,胶体均匀细腻。
2.4.2 纳米粒凝胶的pH 取样品10 g,加100 mL蒸馏水,搅匀,于pH计上依法测定,3批样品的pH分别为7.07、7.23、7.37,符合相关规定。
3 甘草次酸的测定
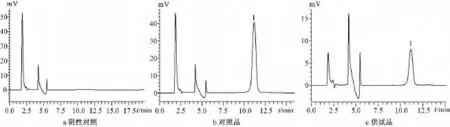
图4 甘草次酸的高效液相色谱图Fig.4 HPLC chromatograms of glycyrrhetinic acid
3.1 色谱条件[15]Shim-pack VP-ODS C18色谱柱(150 mm ×4.6 mm,5μm);流动相为甲醇-水-冰醋酸 (89∶10∶1);体积流量为0.5 mL/min;检测波长为250 nm,进样量为20 μL,柱温为30℃。
3.2 溶液的制备
3.2.1 对照品贮备液的配制 精密称定甘草次酸对照品10.0 mg,置于50 mL量瓶中,甲醇溶解并定容,得甘草次酸贮备液。
3.2.2 标准曲线的制备 精密吸取上述贮备液0.1、0.2、1.0、2.0、4.0、8.0 mL,分别置于20 mL量瓶中并用甲醇定容,得到对照品系列质量浓度 (C)的溶液。按“3.1”项下色谱条件分别进样,记录峰面积 (A)。以A对C进行回归,回归方程:A=52 312C-11 061.9(r=0.999 4),在1.0~80 μg/mL范围内呈良好线性关系。
3.2.3 精密度试验 精密吸取同一供试品溶液20 μL重复进样5次,测定指标峰峰面积,RSD为1.95%,表明仪器精密度良好。
3.2.4 稳定性实验 精密吸取同一供试品溶液20 μL,分别于0、2、4、10、24 h进样,测定甘草次酸色谱峰面积,RSD为1.38%,表明供试品溶液在24 h内稳定性良好。
3.2.5 方法专属性考察 精密吸取用空白接受液浸泡过12 h的小鼠皮的阴性对照溶液1 mL,经0.22 μm微孔滤膜过滤后按上述色谱条件进样分析。由图4可以看出鼠皮溶出物对甘草次酸测定无干扰。3.2.6 回收率试验 分别向已知量的渗透液样品中加入一定量甘草次酸对照品溶液,用流动相加至10 mL混匀后,精密吸取20 μL注入液相色谱仪,测定指标峰峰面积,利用标准曲线计算甘草次酸量,并计算加样回收率。结果甘草次酸平均加样回收率为95.69%,其RSD为0.89%。见表1。

表1 回收率试验结果 (n=9)Tab.1 Results of recovery tests(n=9)
4 甘草次酸固体脂质纳米粒凝胶体外经皮渗透实验
4.1 实验皮肤的制备 取体质量合格的健康昆明种小鼠,脱颈处死,剥取腹部皮肤,剪去毛发及剔除皮下脂肪,用蒸馏水冲洗干净,并用生理盐水冲洗至无浑浊为止,备用 (皮肤用铝箔包裹,置于低温冰箱中保存,试验时取出,室温下自然解冻后使用)。
4.2 经皮渗透实验 取出处理好的小鼠皮肤置于Franz立式扩散池上 (有效扩散面积为3.14 cm2),将角质层朝向供给池,真皮层朝向接受液,将多余的皮肤剪去,固定装置。在接收池中注入19.0 mL磷酸盐缓冲液 (pH 7.3)为接收液,将接收池置于磁力搅拌器上,350 r/min搅拌。另取一套Franz扩散池装置,同法操作,将不含药物的固体脂质纳米粒凝胶空白基质紧贴于离体鼠皮角质层一侧,作为空白对照。分别于规定的时间点0.5、2、4、6、8、12、24 h取样0.5 mL,并及时补充等量新鲜接收液。提取液经0.22 μm微孔滤膜过滤后,取20 μL注入高效液相色谱仪,测定峰面积,代入标准曲线求出药物质量浓度,计算累积渗透药量。每组平行操作3次,平均累计渗透量见图5。小鼠皮肤经过24 h透皮给药试验后,取下后肉眼观看到皮肤表面的纹理较之前清晰可见,再于显微镜下观察未发现破损现象。
根据每个取样点样品的质量浓度,依下式计算皮肤单位面积累积渗透量Q:

式中Q为累积渗透量;Cn和Ci分别为第n、i个取样点测得的药物质量浓度 (g/mL),V和V0分别为接收池体积和取样体积 (mL),A为渗透面积(cm2)。
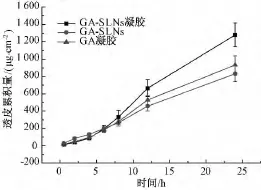
图5 甘草次酸3种剂型透皮累积量 (n=3,±s)Fig.5 Accumulated permeation amount of three glycyrrhetinic acid formulations(n=3,±s)
由图可知甘草次酸固体脂质纳米粒凝胶24 h累计透过量为1 278.4 μg/(cm2·h),甘草次酸水凝胶24 h累计透过量为830.7 μg/(cm2·h),甘草次酸固体脂质纳米粒24 h的累积透过量为931.5 μg/(cm2·h);甘草次酸固体脂质纳米粒凝胶24 h累计透过量较甘草次酸水凝胶提高了30%,较甘草次酸固体脂质纳米粒提高了66%。
5 讨论
固体脂质纳米粒的制备方法有溶剂扩散法、微乳法、高压乳匀法、溶剂乳化挥发法、薄膜-超声分散法[16]。本实验采用微乳液法制备甘草次酸固体脂质纳米甘草次酸固体脂质纳米粒。在制备过程中,磷脂的用量对固体脂质纳米粒的稳定性和粒径均有很大的影响,当磷脂用量超过1.0 g时容易产生沉淀而分层且粒径会增大;脂质材料在乳化时乳化的温度应控制在60~80℃之间,在这个范围内温度越高纳米粒的粒径越小,当温度过高时会引起磷脂或是某些药物的降解,甚至造成混悬液的黏度过大不易洗脱,从而影响制备的固体脂质纳米粒的质量和载药量。高压乳化的压力和循环次数对粒径也有比较大的影响,粒径会随压力增大而变小,在60 MPa压力下,循环6次后粒径基本不随循环次数增加而减小。
由透皮实验的结果可知24 h内甘草次酸固体脂质纳米粒凝胶的总累积透过量相对于甘草次酸固体脂质纳米粒提高了66%,相对于甘草次酸gel总透皮累积量提高了30%。本实验表明,甘草次酸固体脂质纳米粒凝胶与凝胶相比,不仅能增加甘草次酸的透皮速率,提高了药物的生物利用度,甘草次酸固体脂质纳米粒凝胶制剂有希望成为甘草次酸透皮给药的新型制剂。
[1]韩腾飞,闫菁华,豆婧婧,等.甘草次酸醇质体的制备研究[J].西北药学杂志,2012,27(1):64-66.
[2]程 怡,郭波红,林绿萍.甘草次酸脂质体的包封率测定和体外释放度考察[J].广州中医药大学学报,2010,27(4):384-388.
[3]王 森,闫 明,严新安,等.pH对甘草次酸经皮渗透性的影响[J].华西药学杂志,2012,27(2):162-165.
[4]秦宗玲,袁 易.氟比洛芬醇质体凝胶的制备及质量控制[J].中国药房,2007,18(19):1484-1485.
[5]忻志鸣,叶根深.纳米粒制剂研究进展[J].中国药业,2010,19(16):15-16.
[6]Mei Zhinan,Chen Huabing,Weng Ting,et al.Solid lipid nanoparticle and microemulsion for topical delivery of triptolide[J].Eur J Pharm Biopharm,2003,56(2):189-196.
[7]Muller R H,Mader K,Gohla S.Solid lipid nanoparticles(SLN)for controlled drug delivery a review of the state of the art[J].Eur J Pharm Biopharm,2000,50(1):161-177.
[8]田 洁,逄秀娟,吴冬冬,等.顺铂固体脂质纳米粒的制备及其在大鼠体内的分布[J].中国药剂学杂志,2008,6(4):141-142.
[9]熊清平,张强华,石莹莹,等.白藜芦醇固体脂质纳米粒体外抗肝癌HepG2细胞的作用研究[J].亚太传统医药,2011,7(9):12-13.
[10]王 朝,黄绳武.凝胶剂研究进展 [J].医药导报,2010,29(2):223-228.
[11]刘晓昱,饶跃峰,梁文权.炔雌醇醇质体凝胶的经皮渗透研究[J].中国药学杂志,2006,41(4):284-286.
[12]徐 敏,梁文权.α细辛醚凝胶贴剂的体外大鼠皮肤渗透动力学[J].中国临床药学杂志,2006,15(5):318-320.
[13]宁玉明,梁文权.白头翁素凝胶剂的体外经皮渗透及小鼠抗炎试验[J].中国医院药学杂志,2006,26(12):1487-1489.
[14]刘丽宏,辛艳茹,梁冬梅,等.氯麻鼻用凝胶剂 的制备和质量控制[J].解放军药学学报,2007,23(2):140-142.
[15]闫菁华,豆婧婧,等.甘草次酸醇质体水凝胶贴剂的制备与透皮给药研究[J].第二军医大学学报,2011,32(7):763-766.
[16]冯炜玮,陈志伟.固体脂质体纳米粒制备方法的研究进展[J].中国医药生物技术,2011,6(3):218-221.
Preparation and transdermal delivery of glycyrrhetinic acid loaded solid lipid nanoparticles gel
SONG Yan-li, XU Kun, HAN Teng-fei, LI Sha-sha, WEI Hong-hua, CHENG Liang,HAO Bao-hua*
(College of Life Science,Northwest University,Xi’an 710069,China)
glycyrrhetinic acid;solid lipid nanoparticles;gel;transdermal delivery
R944
A
1001-1528(2014)05-0952-05
10.3969/j.issn.1001-1528.2014.05.015
2013-04-12
国家重大新药创制科技重大专项 (2009ZX09502-019)
宋艳丽 (1986—),女,硕士生,研究方向:中药经皮给药。Tel:18192323192,Email:songyanli0402@sina.com
*通信作者:郝保华,男,教授,研究方向:中医基础理论和中药新剂型给药系统。Email:haobaohua@126.com
