去甲基化机制在腺苷及同型半胱氨酸诱导肝癌HepG2细胞凋亡中的作用
2014-05-15项梦琦刘丽璇周小涛陈沛锐郭益添叶艳清蒲泽锦吴灵飞
项梦琦,刘丽璇,邓 巍,周小涛,陈沛锐,郭益添,叶艳清,蒲泽锦,吴灵飞
(汕头大学医学院第二附属医院消化内科,广东汕头 515041)
原发性肝癌是临床常见的恶性肿瘤,其特点是恶性程度高,易复发转移,致死率在肿瘤中排名第3位。目前仅早期发现的病例可通过手术切除而治愈,大多数进展期肝癌则因对常规化疗药物耐药而疗效不佳,因而寻找新的治疗方法增加抗癌效果尤为重要。核苷类物质如腺苷(adenosine,ADO)的细胞毒性作用近年受到关注,它可诱导多种实体与血液肿瘤细胞凋亡,是有前景的抗癌药物[1]。我们前期研究证实,ADO通过膜转运体进入细胞内发挥细胞毒性作用,其机制与内质网应激(endoplasmic reticulum stress,ERS)凋亡途径有关[2-3]。但是 ADO触发凋亡的机制仍不清楚。目前,表观遗传学的研究表明,去甲基化作用可活化抑癌基因如p53而诱导癌细胞凋亡[4]。生理条件下,ADO可与同型半胱氨酸(homocysteine,HCY)合成S-腺苷同型半胱氨酸(s-adenosylhomocysteine,SAH)而降低细胞内的甲基化水平[5]。有文献证实,体外联合使用 ADO及HCY可导致细胞内SAH水平增高,且比单独使用ADO更为明显[6]。为了探讨细胞内甲基化代谢是否参与了ADO细胞毒性作用,我们采用ADO联合HCY作用肝癌HepG2细胞,另外,给予DNA甲基化酶(DNA methyltransferases,DNMTs)特异性抑制剂5-氮杂-2′-脱氧胞苷(5-aza-2′-deoxycytidine,5-Aza-CdR)作为对照,观察对肝癌HepG2细胞生长的影响并探讨其作用机制。
1 材料与方法
1.1 药物和试剂5-氮杂-2′-脱氧胞苷、同型半胱氨酸购自美国Sigma;腺苷购自美国Amerco;DMEM培养基及胎牛血清(Gibco,美国);兔抗人β肌动蛋白、caspase-3、MDM-2、p53抗体、山羊抗兔多克隆二抗(Santa Cruz,美国);caspase-8、caspase-9抗体(Cell Signaling\CST,美国);Cytochrome C(Abcam,英国);CCK8试剂盒购自日本同仁化工;Annexin VFITC/PI Apoptosis Detection Kit购自 Calbiochem;TRNzol总RNA提取试剂盒、RNA逆转录试剂盒及SYBR Premix Ex Taq购自日本TaKaRa。
1.2 细胞培养人肝癌HepG2细胞于含10%的胎牛血清的DMEM培养基,37℃、5%CO2恒温箱培养,呈对数增长期时以0.25%胰酶消化传代。
1.3 CCK-8测定细胞存活率取100μl细胞悬液(含1×105·L-1HepG2细胞)接种于96孔板中培养 24 h,不同剂量 ADO联合 HCY(1.0、2.0、4.0 mmol·L-1)作用肝癌 HepG2细胞6、12或 24 h。5-Aza-CdR(5.0、10.0、20.0μmol·L-1)作用肝癌HepG2细胞24、48或72 h。同时设无药物处理对照组,每组设5个复孔。每孔加100μl(含10%体积CCK8试剂)的DMEM培养基,培养箱内避光培养1 h,酶联免疫检测仪检测570 nm波长的吸光度值(absorbance,A)。实验重复3次。细胞存活率/%=(A实验组-A对照组)/A对照组×100%。
1.4 AnnexinV-FITC/PI双染检测细胞凋亡取2 ml细胞悬液(含3×105·L-1HepG2细胞)接种于6孔板中培养24 h,分阴性对照组、空白对照组和药物处理组。处理组分别加入ADO及HCY 1.0、2.0或4.0 mmol·L-1培养24 h,不含 EDTA的胰酶消化收集细胞,4℃预冷PBS洗细胞3次,取0.5 ml细胞悬液(含1.25μl Annexin V-FITC),室温避光孵育15 min,离心后去上清,0.5 ml缓冲液重悬细胞沉淀,加入10μl PI染液,流式细胞仪上机检测。
1.5 罗丹明123染色测定线粒体膜电位(MMP,ΔΨ)罗丹明123(Rho123)使用前用DMSO配制,使其终浓度为5.0 mmol·L-1,-20℃避光保存。取2 ml细胞悬液(含3×105·L-1HepG2细胞)接种于6孔板中,不同浓度的ADO联合HCY(1.0、2.0、4.0mmol-1)处理24 h后收集细胞,PBS漂洗3次,取0.5 ml细胞悬液(含1×105细胞)于1 ml EP管中;加入终浓度为5.0μmol·L-1的染料于室温避光37℃孵育30 min。PBS洗涤细胞以除去游离的探针。0.5 ml PBS重悬细胞沉淀,流式细胞仪上机检测。激发波长488 nm,发射波长515~575 nm。
1.6 实时荧光定量PCR(RT-qPCR)检测m RNA的表达取2 ml细胞悬液(含3×105·L-1HepG2细胞)接种于6孔板中,HCY及 ADO 1.0 mmol·L-1培养6 h。5-Aza-CdR 10.0μmol·L-1培养72 h。分别收集细胞,TRIzol提取细胞总RNA,测RNA含量及纯度后反转录成cDNA为模板。荧光定量PCR引物应用premier 5.0及oligo 6.0进行设计,扩增片段长度在300 bp以下(引物序列见Tab 1)。20μl的反应体系,应用两步法PCR扩增标准程序,7500 System SDS软件分析目的基因相对表达量(以18S为内参照)。采用2-ΔΔCt法进行目的基因mRNA相对表达水平分析。
1.7 W estern免疫印迹法测定蛋白表达情况取2 ml细胞悬液(含3×105·L-1HepG2细胞)接种于6孔板中,HCY+ADO 1.0 mmol·L-1培养6 h。分别加入适量含PMSF的RAPI裂解液,于冰上充分裂解,收集细胞;4℃下离心15 min;上清检测蛋白浓度。10%的SDS聚丙烯酰胺凝胶电泳分离蛋白后转至PVDF膜。10%脱脂牛奶室温30 min,然后分别加入抗 caspase-3(1∶1 000)、caspase-8(1∶1 000)、caspase-9(1∶1 000)、MDM-2(1∶1 000)、p53(1∶500)、Cytochrome C(1∶1 000)抗体 4℃过夜,充分洗膜后分别加入二抗 (1∶3 000)室温孵育1~2 h。用ECL法发光,于暗室曝光。Quantity One进行半定量分析。

Tab 1 Primers for Quantitative Real-Time PCR
1.8 统计学分析实验数据以±s表示,采用SPSS 15.0统计软件进行统计学处理,两组间均数比较采用独立样本t检验,多组间采用单因素方差分析(One-way ANOVA)。
2 结果
2.1 ADO联合HCY对HepG2细胞生长抑制作用单独使用HCY对HepG2细胞作用24 h对其生长无明显影响。ADO随着浓度增加,细胞生长受到抑制;ADO联合HCY作用,细胞生长抑制更为明显,并呈量效与时效依从性(Fig 1A、B)。由于较小浓度腺苷在细胞内可被腺苷脱氢酶迅速代谢[6],因此我们选择低剂量ADO与 HCY联合用药(1.0 mmol·L-1)6 h作为下一步的研究。不同浓度5-Aza-CdR(5.0、10.0、20.0μmol·L-1)作用 HepG2细胞72 h时,细胞出现明显生长抑制。因此,我们选10.0μmol·L-1剂量、72 h进行下一步的研究。

Fig 1 Effects of ADO and HCY or 5-Aza-CdR on the cell viability
2.2 ADO联合HCY对HepG2细胞凋亡的作用ADO联合 HCY 1.0、2.0、4.0 mmol·L-1处理细胞24 h,凋亡率分别为(18.63±1.25)%,(29.42±2.37)%,(42.47±3.09)%,对照组为(1.30±0.82)%,药物处理组与对照组相比,差异均有显著性(P<0.05,n=3)。见 Fig 2。
2.3 ADO联合HCY对线粒体膜电位的影响ADO联合 HCY 1.0、2.0、4.0 mmol·L-1处理细胞24 h,ΔΨ由空白对照组(674.15±82.8)%,随浓度增高荧光强度逐渐下降,分别为(428.38±54.5)%、(297.78±30.5)%、(74.45±5.73)%,与空白对照组相比,差异均有显著性(P<0.01)。见Fig 3。
2.4 ADO联合HCY对HepG2细胞凋亡及甲基化相关因子m RNA表达的影响ADO联合HCY(浓度各为 1.0 mmol·L-1)处理细胞 6 h后,caspase-3、caspase-8、caspase-9、p53、Cytochrome C mRNA表达增加,MDM-2 mRNA表达下降。5-Aza-CdR 10.0μmol·L-1处理细胞 72 h或者 ADO联合HCY处理细胞 6 h,DNA甲基化酶 DNMT1、DNMT3a、DNMT3b mRNA表达下降,与对照组相比,差异均有显著性 (P<0.05,)。见Fig 4。
2.5 ADO联合HCY对HepG2细胞凋亡相关因子的蛋白表达的影响ADO联合HCY(浓度各为1.0 mmol·L-1)处理细胞6 h后,caspase-3、caspase-8、caspase-9、p53、Cytochrome C 蛋白表达增加,MDM-2蛋白表达下降,与对照组相比,差异均有显著性 (P<0.05)。见Fig 5。
3 讨论

Fig 2 Effects of ADO and HCY treatment on apoptosis of HepG2 cells
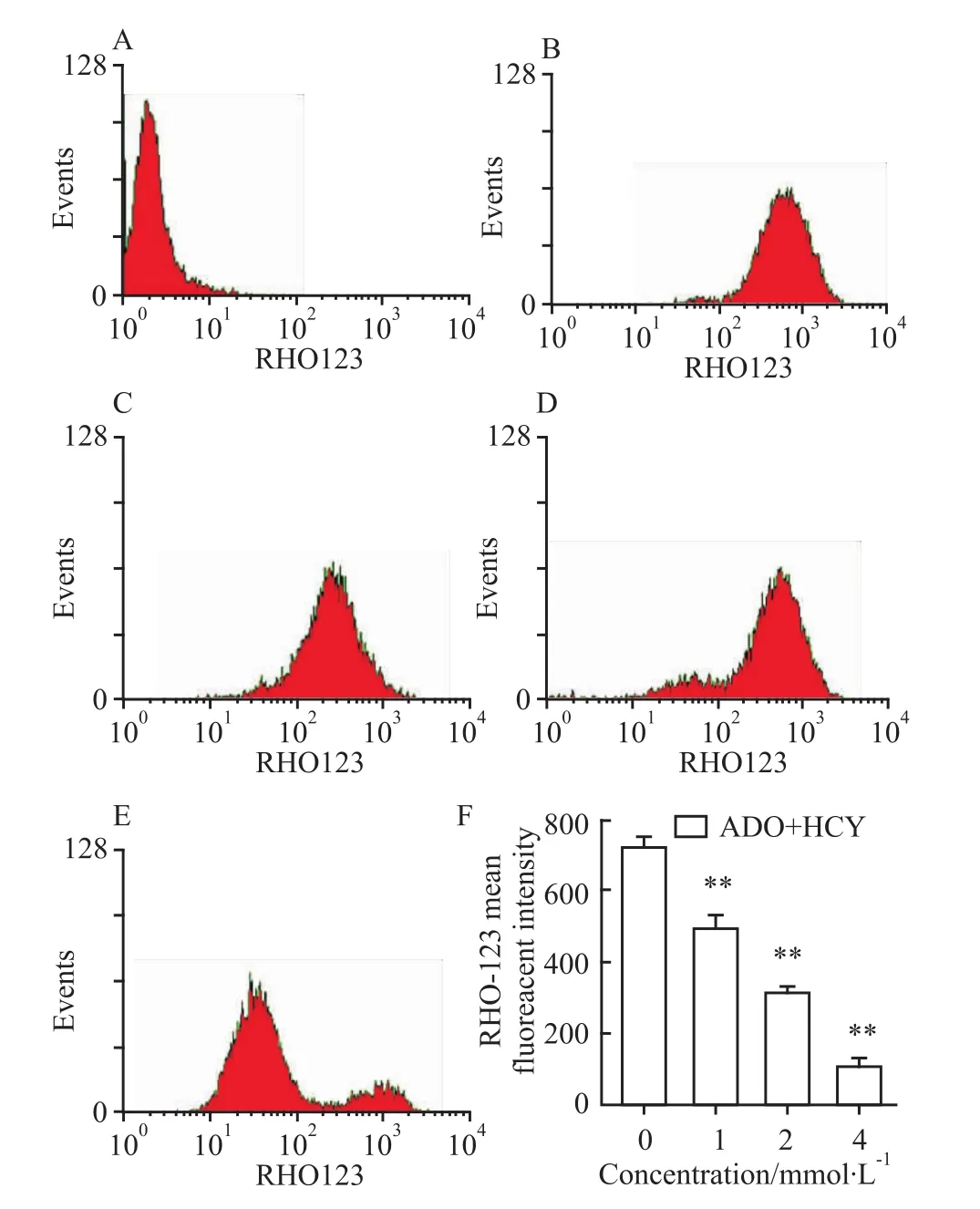
Fig 3 Effects of combination treatment with ADO and HCY on m itochondrialmembrane potentials(MMP,ΔΨ)
ADO在细胞内有3条不同的代谢途径。ADO可通过脱氨代谢途径生成肌苷,通过磷酸代谢途径合成一磷酸腺苷(AMP)或与细胞内 HCY合成SAH。我们既往的研究证实,腺苷的脱氨代谢途径与其细胞毒作用无关[7]。近年随着表观遗传学DNA甲基化研究的深入,ADO对胞内甲基化代谢的影响受到关注,有研究表明ADO可与HCY合成SAH而降低细胞内的甲基化水平[5-6],ADO发挥抗癌作用是否与DNA去甲基化作用有关引起了我们的兴趣。生理状态下,在蛋氨酸循环中,S-腺苷蛋氨酸(SAM)转甲基后生成 S-腺苷同型半胱氨酸(SAH),SAH则在 SAH水解酶作用下快速脱去ADO而生成HCY以保证细胞内正常的甲基化水平,这是一可逆反应。ADO亦可与HCY合成SAH,ADO及HCY水平增加可导致上述反应向合成SAH的方向进行。Hermes等[6]研究证实ADO及其类似物ADOX均对SAH水解酶有抑制作用,并可导致多种凋亡相关基因上调,显示ADO抑制SAH水解酶参与了凋亡的诱导。另有文献报道[8],SAH具有抑制DNMTs的作用。本实验结果表明,ADO联合HCY处理HepG2细胞后,可明显抑制DNMTs的活性及细胞生长,并诱导了细胞凋亡,表明ADO联合HCY的细胞毒性作用与降低细胞内的甲基化水平有关。
表观遗传学研究表明,肿瘤的发生及进展与细胞内DNA总体的低甲基化及局部区域的高甲基化、尤其是抑癌基因的高甲基化有关[9]。DNA甲基化酶(包括DNMT1、DNMAT3a以及DNMT3b)已被证实在肝癌HepG2细胞中常出现高表达并导致多种抑癌基因失活[10]。表观遗传学变化中某些关键基因启动子区域的甲基化状态可通过药物处理而改变并在临床试验中成为药物靶点,采用DNA甲基化酶抑制剂(5-Aza-CdR)已在多种肿瘤中证实具有抗癌作用并与活化抑癌基因有关[11]。本实验中ADO联合HCY可明显抑制DNMTs活性并活化抑癌基因p53,具有与5-Aza-CdR一致的效果,显示ADO联合HCY可通过去甲基化、活化抑癌基因p53发挥抗癌作用。在肝癌HepG2细胞中,由于p53是野生型,它可通过多种途径触发细胞凋亡[9],因此,p53在肝癌治疗研究中的作用尤为重要。新近有文献报道,MDM-2可通过反馈环对p53的功能进行调节,即MDM-2通过泛素化与p53结合而抑制p53的转录活性[12]。本实验中,我们观察到ADO联合HCY作用HepG2细胞后MDM-2的mRNA及蛋白水平表达量下降、p53表达量增高,支持MDM-2负反馈调节p53的观点[12]。ADO联合 HCY如何降低 MDM-2的具体机制则有待进一步研究。
ADO及HCY联合用药后,HepG2细胞线粒体膜电位降低,细胞色素C、caspase-9及caspase-3mRNA及蛋白表达明显增加(Fig 4-5)。目前认为,线粒体凋亡通路是通过凋亡因子诱发Bcl-2家族(主要是Bax)的活化,致使线粒体膜损伤、膜电位下降而释放细胞色素C,细胞色素C与Apaf-1结合可活化caspase-9并最终激活效应因子caspase-3触发凋亡[13]。而p53-bax线粒体凋亡通路中,许多基因均有潜在的甲基化位点,并可因甲基化而失活,结果丧失了该通路的抗肿瘤功能[14]。本实验中,我们观察到ADO及HCY联合用药后通过去甲基化作用激活了p53。结合我们既往研究结果,ADO作用后可引起 Bcl-2水平下降,Bax和 Bad表达升高[2,15],以及线粒体膜电位降低、细胞色素C释放及caspase-9水平增加,因此,我们认为线粒体通路参与了ADO的细胞毒性作用。

Fig 4 Effects of combination treatment with ADO and HCY on m RNA relative expressions of caspase-3,caspase-8,caspase-9,MDM-2,p53,Cytochrome C(A),5-Aza-CdR or ADO and HCY on m RNA relative expressions of DNMT1,DNMT3a,DNMT3b in HepG2 cells(B)

Fig 5 Effects of ADO and HCY on protein expressions of caspase-3,caspase-8,caspase-9,MDM-2,p53,Cytochrome C in HepG2 cells 0 as control group,1 as ADO and HCY treatment group.*P<0.05,**P<0.01 vs control group.Independent experiments were repeated three times.
此外,ADO与HCY联合作用后,我们还观察到caspase-8 mRNA及蛋白表达明显增加(Fig 4、5)。caspase-8是死亡受体凋亡通路中的一个重要蛋白[16],在结构上它的启动子区域有 p53、NF-κB、AP-1,SP-1,IRF-1的结合位点。我们在既往的研究中证实,ADO可引起分子伴侣GRP78及NF-κB表达增加,它们均有保护肿瘤细胞生存的特性,分别可作为肿瘤耐药的第一及第二道重要屏障[2,15]。因此,caspase-8不仅是死亡受体通路的重要分子,而且还可作为选择性的信号转导因子影响 NF-κB的功能[17],显示肿瘤凋亡过程中信号调节的复杂性[4]。在本项研究中,ADO与HCY联合作用后caspase-8、caspase-3 mRNA及蛋白表达增加,并出现细胞生长抑制。因此我们推测死亡受体通路可能亦参与了ADO诱导的细胞凋亡作用。
总之,本研究结果表明,ADO联合HCY可通过抑制甲基转移酶、降低细胞内甲基化代谢作用而活化抑癌基因P53并诱导肝癌细胞凋亡。其机制与活化线粒体通路有关,死亡受体通路可能也参与了此过程。
参考文献:
[1]Antonioli L,Blandizzi C,Pacher P,HaskóG.Immunity,inflammation and cancer:a leading role for adenosine.[J].Nat Rev Cancer,2013,13(12):842-57.
[2]Wu L F,Guo Y T,Zhang QH,etal.Enhanced antitumor effects of adenoviral-mediated siRNA against GRP78 gene on adenosineinduced apoptosis in human hepatoma HepG2 cells.[J].Int J Mol Sci,2014,15(1):525-44.
[3]叶艳清,李国平,蒲泽锦,等.腺苷通过内质网应激途径诱导HepG2细胞凋亡的研究[J].中国药理学通报,2010,26(5):596-601.
[3]Ye Y Q,LiG P,Pu Z J,etal.The research of adenosine induced HepG2 cell apoptosis through endoplasmic reticulum stress pathway[J].Chin Pharmacol Bull,2010,26(5):596-601.
[4]Qiang W,Jin T,Yang Q,et al.PRIMA-1 Selectively Induces Global DNA Demethylation in p53 Mutant-Type Thyroid Cancer Cells[J].JBiomedical Nanotechnol,2014,10(7):1249-58.
[5]Kalhan S C,Marczewski S E.Methionine,homocysteine,one carbon metabolism and fetal growth[J].Rev Endocr Metab Disord,2012,13(2):109-19.
[6]Hermes M,Osswald H,Kloor D.Role of S-adenosylhomocysteine hydrolase in adenosine-induced apoptosis in HepG2 cells[J].Exp Cell Res,2007,313(2):264-83.
[7]Wu L F,Ye Y Q,Huang GY,et al.Involvement of endoplasmic reticulum stress in adenosine-induced human hepatoma HepG2 cell apoptosis[J].Oncol Rep,2011,26(1):73-9.
[8]Zhang X,Li H,Qiu Q,et al.2,4-Dichlorophenol induces global DNA hypermethylation through the increase of S-adenosylmethionine and the upregulation of DNMTsmRNA in the liver of goldfish Carassius auratus[J].Comp Biochem Physiol C Toxicol Pharmacol,2014,160:54-9.
[9]Leonova K I,Brodsky L,Lipchick B,et al.P53 cooperates with DNA methylation and a suicidal interferon response tomaintain epigenetic silencing of repeats and noncoding RNAs.[J].Proc Natl Acad Sci USA,2013,110(1):E89-98.
[10]Oh B K,Kim H,Park H J,et al.DNAmethyltransferase expression and DNA methylation in human hepatocellular carcinoma and their clinicopathological correlation[J].Int JMolMed,2007,20(1):65-73.
[11]Liu J,Xie Y S,Wang F L,et al.Cytotoxicity of 5-Aza-2′-deoxycytidine against gastric cancer involves DNA damage in an ATMP53 dependent signaling pathway and demethylation of P16 INK4A[J].Biomed Pharmacother,2013,67(1):78-87.
[12]Clegg H V,Itahana Y,Itahana K,etal.Mdm2 RINGmutation enhances p53 transcriptional activity and p53-p300 interaction[J].PloS one,2012,7(5):e38212
[13]Mohamad N,Gutiérrez A,NúñezM,etal.Mitochondrial apoptotic pathways[J].Biocell,2005,29(2):149-61.
[14]Schuler M,Green D R.Mechanisms of p53-dependent apoptosis[J].Biochem Soc Trans,2001,29(6):684-8.
[15]Wu L F,Li G P,Su JD,et al.Involvement of NF-κB activation in the apoptosis induced by extracellular adenosine in human hepatocellular carcinoma HepG2 cells[J].Biochem Cell Biol,2010,88(4):705-14.
[16]Liedtke C,Groger N,Manns M P,Trautwein C.The human caspase-8 promoter sustains basal activity through SP1 and ETS-like transcription factors and can be upregulated by a p53-dependentmechanism[J].JBiol Chem,2003,278(30):27593-604.
[17]Koenig A,Buskiewicz I A,Fortner K A,et al.The c-FLIPL cleavage product p43FLIP promotes activation of extracellular signal-regulated kinase(ERK),nuclear factorκB(NF-κB),and caspase-8 and T cell survival[J].J Biol Chem,2014,289(2):1183-91.
