LPS/D-GalN诱导小鼠急性肝损伤模型的建立
2014-05-10吴小红郭彦刘晨风2高同同于虹孙世惠周育森
吴小红,郭彦,刘晨风2,,高同同,于虹,孙世惠,周育森
(1.病原微生物生物安全国家重点实验室,北京微生物流行病研究所,北京 100071; 2.安徽医科大学北京微生物流行病研究所,合肥 230032)
研究报告
LPS/D-GalN诱导小鼠急性肝损伤模型的建立
吴小红1,郭彦1,刘晨风2,1,高同同1,于虹1,孙世惠1,周育森1
(1.病原微生物生物安全国家重点实验室,北京微生物流行病研究所,北京 100071; 2.安徽医科大学北京微生物流行病研究所,合肥 230032)
目的建立脂多糖(lipopolysaccharide,LPS)/D-氨基半乳糖(D-galactosamine,D-GalN)诱导小鼠急性肝损伤模型。方法40只雌性C57BL/6小鼠用于观察8种不同LPS与D-GalN剂量配比联合刺激后小鼠存活时间,以确定模型建立的最佳剂量。使用腹腔注射最佳剂量染毒32只雌性C57BL/6小鼠,分别在0、1、4、8 h处死,每组8只,0 h注射相同剂量生理盐水作为对照。观察染毒后小鼠肝组织病理损伤,检测血清中ALT及炎症因子IL-6、MCP-1和TNF-α表达水平变化。结果通过观察小鼠存活时间,确定腹腔注射最佳染毒剂量为LPS(2.5 mg/ kg)/D-GalN(0.3 g/kg);小鼠染毒后肝组织呈进程性病变,最终发展为肝脏弥漫性坏死,肝细胞核崩解。与对照组相比,血清ALT显著升高(P<0.001),IL-6、MCP-1、TNF-α均在1 h后达到最高水平(P<0.001),然后持续下降。结论 成功建立LPS/D-GaIN诱导小鼠急性肝损伤模型,为探索急性肝损伤的致病机制以及药物干预治疗提供有效的动物模型。
内毒素;D-氨基半乳糖;急性肝损伤;炎症;小鼠
急性肝损伤是目前临床上常见的一种疾病,如果未得到及时的控制和治疗,将会发展为重症肝炎等不可逆性疾病,临床报道显示重症肝炎介导的病死率非常高[1]。而外界肝炎病毒、化学性毒物或药物、酒精及放射性刺激因素均可引起急性肝损伤[2-4],其病情发展迅速、凶险,临床典型特征表现为肝功能衰竭,肝组织中大量肝细胞坏死和凋亡[5-6]。目前认为,急性肝损伤是肝实质细胞严重急性损害的结果[7]。虽然开展了大量的研究工作探讨其致病机制,但是目前尚未完全清楚。因此,建立有效的急性肝损伤动物模型是探索其损伤机制以及药物防治方法等的前提。本实验通过建立内毒素伴随D-氨基半乳糖(LPS/D-GalN)诱导小鼠内毒素性急性肝损伤模型,以期能够建立有效并且稳定的急性肝损伤模型,为探索急性肝损伤致病机制等研究奠定基础。
1 材料与方法
1.1 材料
1.1.1 材料及试剂
LPS/D-GalN购自Sigma公司,苏木素、伊红、中性树胶等HE染色技术相关耗材均购自北京中杉金桥生物科技有限公司;小鼠炎症细胞因子检测试剂盒(Cytometric Bead Array(CBA),Mouse Inflammation Kit)购自BD公司,40%甲醛溶液购自上海国药集团化学试剂有限公司,其余试剂均为国产分析纯。1.1.2 实验动物
SPF级雌性C57BL/6小鼠72只,6周龄,体重18~20 g左右,购自军事医学科学院实验动物中心[SCXK(京)2007—0004],所有动物染毒实验均在SPF级屏障室内进行【SYXK(京)2012-0004】。
1.2 方法
1.2.1 动物模型建立
其中40只小鼠随机分为8组,每组5只,腹腔注射不同剂量LPS/D-GalN(注射体积均为每只200 μL,生理盐水溶解,下同)后观察小鼠的存活状态及存活时间,以确定急性肝损伤小鼠模型建立最佳剂量。8组小鼠注射毒物剂量分别为:LPS(5 mg/ kg)/D-GalN(0.5 g/kg)、LPS(5 mg/kg)/D-GalN (0.4 g/kg)、LPS(4 mg/kg)/D-GalN(0.4 g/kg)、 LPS(4 mg/kg)/D-GalN(0.3 g/kg)、LPS(2.5 mg/ kg)/D-GalN(0.3 g/kg)、LPS(2.5 mg/kg)/D-GalN (0.25 g/kg)、LPS(2.5 mg/kg)/D-GalN(0.2 g/ kg)、LPS(2.5mg/kg)/D-GalN(0.15 g/kg)。确定最佳剂量后,另32只小鼠随机分为对照0 h、染毒1 h、染毒4 h、染毒8 h共4组,每组8只。腹腔注射200μL最佳剂量LPS/D-GalN生理盐水溶液,分别在染毒1 h、4 h、8 h后处死,对照组注射相同剂量生理盐水。小鼠处死后收集血清用于炎症因子检测,收集肝组织固定于4%中性甲醛溶液中用于病理学观察。
1.2.2 小鼠染毒后存活时间及状态观察
40只小鼠随机分为8组,按8种不同剂量LPS和D-GalN染毒后,观察小鼠染毒后生命状态,记录小鼠存活时间,选择最佳LPS和D-GalN剂量。
1.2.3 肝组织病理学观察
小鼠肝组织分别经过4%甲醛固定、自来水复水、酒精梯度脱水、石蜡包埋切片(5μm),苏木素-伊红(HE)染色,封片后光学显微镜观察并拍照。
1.2.4 血清ALT的检测
收集小鼠血液,1500 g离心20 min后收集小鼠血清,取50μL血清送至军事医学科学院动物中心生化分析室,用Beckman全自动生化分析仪CX5检测。
1.2.5 血清炎症细胞因子的检测
检测血清中炎症因子IL-6、MCP-1、TNF-α表达水平,使用BD公司的CBA Mouse Inflammation Kit,混匀小鼠炎症因子捕获珠、分别加入各炎症因子标准品,待测小鼠血清样品,炎症因子PE指示剂,避光混匀室温孵育2 h,流式细胞仪检测并分析,具体方法按BD公司小鼠炎症因子检测试剂盒说明书进行操作。
1.2.6 统计学分析
数据处理采用GraphPad Prism 5软件进行统计分析。组间比较采用t检验,P<0.05为具有统计学差异,P<0.01具有显著统计学差异,P<0.001具有极显著统计学差异。
2 结果
2.1 小鼠急性肝损伤模型中LPS和D-GalN最佳使用浓度的探索
观察注射不同剂量浓度LPS/D-GalN的8组C57BL/6小鼠,并记录死亡时间。如图1所示,在全部致死的 5组中,其中 LPS(5mg/kg)/D-GalN (0.5g/kg)、LPS(5 mg/kg)/D-GalN(0.4 g/kg)、LPS(4mg/kg)/D-GalN(0.4 g/kg)、LPS(4 mg/ kg)/D-GalN(0.3 g/kg)这4组C57BL/6小鼠在4~5 h内全部死亡,病程发展太快,不适合用于建立一个急性肝损伤模型的病变过程。因此,在后续实验中应选择毒物用量既能建立有效的急性肝损伤的模型,又能在染毒后能够反映病程变化的进程。对比发现,采用在8~12h全部致死的剂量,即LPS (2.5 mg/kg)/D-GalN(0.3 g/kg)作为最佳染毒剂量。
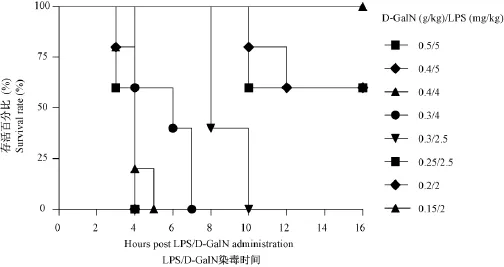
图1 不同配比剂量LPS和D-GalN染毒C57BL/6小鼠存活率观察Fig.1 Survival rates of the C57BL/6 mice after LPS/D-GalN administration in different doses
2.2 LPS/D-GalN诱导C57BL/6小鼠内毒性急性肝损伤模型病理学变化
为了观察毒物对小鼠肝脏的病理损伤,我们使用HE染色的方法观察了肝组织病理变化,结果如图2所示,C57BL/6小鼠随着染毒时间的延长,肝组织石蜡切片HE染色显示肝组织呈现急性内毒素性肝炎急性肝损伤进程性病变。正常C57BL/6小鼠(见图2A)肝脏组织形态正常,肝小叶结构清楚。LPS/D-GalN染毒1 h后(见图2B)肝小叶结构清楚,枯否细胞增多,出现小血管溶血。LPS/D-GalN染毒4 h后(见图2C)肝组织实质灶性坏死,大面积出血。LPS/D-GalN染毒8 h后(见图2D)肝小叶结构紊乱,肝实质大面积灶性坏死,细胞核崩解,肝管与血管严重出血(图2见彩插3)。
2.3 LPS/D-GalN致C57BL/6小鼠内毒性急性肝损伤模型血清ALT水平变化
从图3A可以看出,C57BL/6小鼠血清ALT水平显著性增高,并且ALT水平增高具有时间依赖性,并在8h后达到最高水平(P<0.001)。
2.4 C57BL/6小鼠内毒素性急性肝损伤模型血清炎症细胞因子水平变化
从图3可以看出:在小鼠急性肝损伤模型中, C57BL/6小鼠内毒素性急性肝损伤时其血清中炎症因子IL-6、MCP-1及TNF-α水平均呈先升高再降低的趋势,在早期(1 h时)即达峰值(均P<0.001),随后急剧降低。
3 讨论
急性肝损伤是急性肝衰竭的基础,严重或持续的肝损伤,最终导致急性重症肝炎或爆发性肝炎而导致死亡[1-3]。引起急性肝损伤的原因很多,主要有病毒感染,用药不当,乙醇摄入过多及食入有毒食物或放射性损伤等[2-4]。急性重型肝炎的发病机制较为复杂,迄今尚不清楚。建立稳定而有效的急性肝损伤动物模型是探索急性重型肝炎致病机制研究的重要环节。
目前建立急性肝损伤模型的方法主要有病毒性模型、免疫性模型及化学性模型[8-10]。其中化学性模型又以四氯化碳和内毒素致动物急性肝损伤模型较为常用。研究表明,内毒素不仅可以直接或间接造成肝细胞的损伤,而且其诱导的肝细胞坏死在重症肝炎的发展进程中起重要作用[10]。脂多糖(lipopolysaccharide,LPS)是内毒素的主要毒性作用成分,可介导炎症因子破坏血管内皮的完整性,导致肝细胞的凋亡和坏死,同时引起肝脏的损伤及出血[11-12]。D-氨基半乳糖(D-galactosamine,D-GalN)是常用的化学性肝毒性药物,可快速结合并消耗大量的尿苷酸,影响肝细胞蛋白质、酶等的生成,从而对肝细胞组织造成不可逆的损伤[10-12]。利用LPS/ D-GalN诱导动物发生急性肝损伤,不仅能建立内毒素性急性肝损伤动物模型,同时可用于急性重症肝炎模型的研究[10-12]。但是不同的小鼠品系对不同剂量配比的LPS/D-GalN所产生的病理学损伤及炎症免疫反应存在差异[13-16]。为此,我们探索 LPS和D-GalN的适宜给药剂量,建立稳定的急性肝损伤动物模型,为进一步开展急性肝损伤的损伤机制、生理机制研究及对治疗药物评价奠定基础。
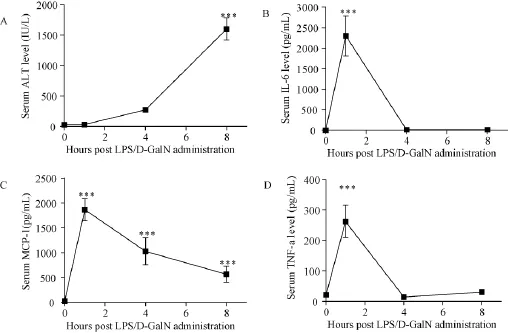
图3 LPS/D-GalN染毒C57BL/6小鼠后血清ALT(A)、IL-6(B)、MCP-1(C)、TNF-α(D)的表达Fig.3 The concentrations of serum ALT(A),IL-6(B),MCP-1(C)and TNF-α (D)in the C57BL/6 mice after LPS/D-GalN administration
病理学研究发现,以LPS/D-GalN染毒小鼠,小鼠肝损伤呈急性渐进式发展。正常C57BL/6小鼠肝小叶结构清楚,随着病程的发展,肝组织由散在的点状灶性坏死,肝细胞胞质疏松发展至弥漫性的大面积出血性坏死,细胞核崩解,无肝小叶正常结构。ALT存在于肝细胞的胞质中,当发生肝组织急性损伤时,肝细胞膜结构破坏,通透性增加,ALT从细胞内到细胞外而进入血液,使血清中ALT水平明显升高,是急性肝损伤的重要的早期检测指标之一。在四氯化碳或病毒诱导的急性肝损伤模型中均能检测到血清中ALT水平急剧升高[9],本研究中ALT在LPS/D-GalN染毒后早期即迅速上调,提示组织的早期即受到一定程度的损伤。
越来越多的研究表明,急性肝损伤的致病机制与体内的免疫应答及免疫调节机制有关,特别是炎症细胞因子的失衡与肝脏损伤密切相关[17-19]。研究发现LPS对肝细胞有直接毒性,可激活Toll样受体4(Toll-like receptor-4,TLR4),TLR4的激活促进了枯否细胞等单核细胞对TNF-α、IL-6等炎症因子的大量表达[20]。IL-6是白介素家族的重要成员,是一种多功能生长因子,在机体炎症、免疫调节等方面发挥重要作用,近年来大量文献报道IL-6与肝脏疾病密切相关[21]。而TNF-а同样与急性肝损伤有非常密切的关系[22-23],研究表明,TNF-α诱导细胞凋亡甚至坏死,刺激内皮细胞表面表达促凝血蛋白C,从而使内皮细胞表面由正常抗凝状态变成促凝状态,有利于血栓形成和急性肝坏死,最后导致肝组织的免疫病理损伤。MCP-1(单核细胞趋化蛋白)是趋化因子家族重要成员之一,属CC类细胞趋化因子,对单核细胞及巨噬细胞和T细胞具有特异性趋化和激活作用。LPS可诱导内皮细胞表达MCP-1的增加[17-19,24]。由本实验的结果可知,在小鼠染毒早期,血清中细胞炎症因子IL-6、TNF-α、MCP-1表达量就迅速升高,这些炎症因子活化后,进一步激活体内的免疫应答,活化体内白介素家族,肿瘤坏死因子家族以及趋化因子家族,以上的级联反应共同导致了体内的“炎症因子风暴”,进而介导了急性肝损伤的恶化,最终引发重症肝炎等不可逆型肝脏疾病。实验中还发现,在LPS染毒后期炎症因子均发生明显下调,可能的原因是由于LPS毒性较大,早期诱发机体强烈的免疫反应,但在4h后造成机体各种巨噬细胞、淋巴细胞的大量坏死,上述细胞坏死后不能将合成的炎症蛋白分泌到胞外或入血,以致使染毒后期血清炎症因子明显下降[22-26]。
综上,从肝组织病理学、血清ALT水平及炎症因子水平等方面来LPS/D-GalN诱导的肝组织损伤符合重症肝炎急性肝损伤发生发展的表现。本实验建立的内毒素性急性肝损伤小鼠模型具有较好的稳定性,易行性。该急性肝损伤实验动物模型的建立,将为进一步开展急性肝损伤的致病机制等研究提供理想的实验动物模型。
(本文图2见彩插3。)
[1] Hoofnagle JH,Carithers RLJ,Shapiro C,et al.Acute hepatic failure:summary of a workshop[J].Hepatology.1995,21: 240-252.
[2] Ichai P,Samuel D.Etiology and prognosis of fulminant hepatitis in adults[J].Liver Transpl,2008,14(Suppl 2):S67-79.
[3] Acharya SK,Batra Y,Hazari S,et al.Etiopathogenesisof acute hepatic failure:eastern versusWestern countries[J].JGastroenterol Hepatol,2002,17(Suppl 3):S268-273.
[4] Stine JG,Lewis JH.Drug-induced liver injury:a summary of recent advances[J].ExpertOpin Drug Metabol Toxicol.2011;7: 875-890.
[5] Kaplowitz N.Mechanisms of liver cell injury[J].JHepatol. 2000,32:39-47.
[6] Rust C,Gores GJ.Apoptosis and liver disease[J].Am JMed. 2000,108:567-574.
[7] Jalan R.Pathophysiological basis of therapy of raised intracranial pressure in acute liver failure[J].Neurochem Int.2005,47: 78-83.
[8] Nakama T,Hirono S,Moriuchi A,et al.Etoposide prevents apoptosis in mouse liver with D-galactosamine/lipopolysaccharideinduced fulminant hepatic failure resulting in reduction of lethality[J].Hepatology.2001,33:1441-1450.
[9] Zhu RZ,Zeng GF,Chen YQ,etal.Oroxylin A accelerates liver regeneration in CCl4-induced acute liver injury mice[J].PloS ONE.2013,8:e71612.
[10] Chastre A,Bélanger M,Beauchesne E,etal.Inflammatory cascades driven by tumor necrosis factor-alpha play a major role in the progression of acute liver failure and its neurological complications[J].PloSONE.2012.7:e49670.
[11] Liu D,Li C,Chen Y,et al.Nuclear import of proinflammatory transcription factors is required for massive liver apoptosis induced by bacterial lipopolysaccharide[J].JBiol Chem,2004, 279:48434-48442.
[12] Fukuda T,Mogami A,Tanaka H,et al.Y-40138,a multiple cytokine production modulator,protects against D-galactosamine and lipopolysaccharide-induced hepatitis[J].Life Sci.2006,79 (9):822-827.
[13] Ikeda T,Abe K,Kuroda N,et al.The inhibition of apoptosis by glycyrrhizin in hepatic injury induced by injection of lipopolysaccharide/D-galactosamine inmice[J].Arch Histol Cytol.2008, 71:163-178.
[14] Zhao J,Kim KD,Yang X,etal.Hyper innate responses in neonates lead to increased morbidity and mortality after infection[J].Proc Natl Acad Sci U SA.2008,105:7528-7533.
[15] Miyanaga K,Yoshioka T,Nakagawa H,et al.Influence ofmurine hepatitis induced by D-(+)-galactosamine hydrochloride and lipopolysaccharide on gene expression of polyethylenimine/ plasmid DNA polyplex[J].Biol Pharm Bull.2008,31:1585-1589.
[16] Zhou BR,Gumenscheimer M,Freudenberg MA,et al.Lethal effect and apoptotic DNA fragmentation in response of D-GalN-treated mice to bacterial LPS can be suppressed by pre-exposure tominute amount of bacterial LPS:dual role of TNF receptor 1[J].World JGastroenterol.2005,11:3398-3404.
[17] Szabo G,Mandrekar P,Dolganiuc A.Innate immune response and hepatic inflammation[J].Semin Liver Dis.2007,27:339 -350.
[18] Wu Z,Han M,Chen T,et al.Acute liver failure:mechanisms of immune-mediated liver injury[J].Liver Int.2010,30:782 -794.
[19] Blazka ME,Wilmer JL,Holladay SD,et al.Role of proinflammatory cytokines in acetaminophen hepatotoxicity[J].Toxicol Appl Pharmacol.1995,133:43-52.
[20] Yan CG,Qing X,Zhou XQ,etal.The expression of toll-like receptor 4 in the endotoxin-induced acute hepatic injury[J]. China J Infect Dis.2004,22:189-192.
[21] Diehl AM.Cytokine regulation of liver injury and repair[J]. Immunol Rev,2000,174:160-171.
[22] Nagaki M,IwaiH,Naiki T,etal.High levels of serum interleukin-10 and tumor necrosis factor-alpha are associated with fatality in fulminant hepatitis[J].J Infect Dis.2000,182:1103-1108.
[23] Streetz K,Leifeld L,Grundmann D,etal.Tumor necrosis factor alpha in the pathogenesis of human and murine fulminanthepatic failure[J].Gastroenterology.2000,119:446-460.
[24] Butterworth RF.Neuroinflammation in acute liver failure:mechanisms and novel therapeutic targets[J].Neurochem Int.2011, 59:830-836.
[25] 陈誉华.医学细胞生物学[M].人民卫生出版社,第四版, 2008:89-124.
[26] 龚非力.医学免疫学[M].科学出版社,第三版,2009:46-61.
Establishment of a mousemodel of acute liver failure induced by LPS/D-GalN
WU Xiao-hong1,GUO Yan1,LIU Chen-feng2,1,GAO Tong-tong1,YU Hong1,SUN Shi-hui1,ZHOU Yu-sen1
(1.State Key Laboratory of Pathogen and Biosecurity,Beijing Institute of Microbiology and Epidemiology,Beijing 100071,China; 2.Beijing Institute of Microbiology and Epidemiology,AnhuiMedical University,Hefei230032)
ObjectiveTo establish amousemodel of acute liver failure induced by lipopolysaccharide/D-galactosamine(LPS/D-GalN).M ethodsThe optimum dose of LPS/D-GalN was determined by i.p.injection of eight different doses of LPS and D-GalN into 40 female C57BL/6 mice and observation of their survival time.Then,32 female C57BL/6 micewere i.p.injected with the optimal dose of LPS/D-GalN and sacrificed at0,1,4,8 hours after the injection,8mice in each group.The controlmice received saline injection.Hepatic changeswere observed by pathology and serum ALT,IL-6,MCP-1 and TNF-αweremeasured by biochemistry or flow cytometry.ResultsLPS(2.5 mg/kg)and D-GalN(0.3 g/kg)were determined as the optimal dose for the establishmentofmousemodel of acute liver injury.Compared with the control group,the hepatocellular damageswere progressing in a positive correlation with the time course after LPS/D-GalN administration.The level of serum ALT was significantly increased after LPS/D-GalN administration(P<0.001).The levels of inflammatory cytokines IL-6,MCP-1 and TNF-αwere increased and reached a peak atone hour after LPS/D-GalN administration and then decreased almost to thatof the control group 8 hours later(P<0.001).ConclusionsThemousemodelof acute liver injury is successfully established by LPS/D-GalN administration,and provide an effective animalmodel for the study of pathogenicmechanisms of acute liver failure and evaluation of therapeutic drugs.
Lipopolysaccharide,LPS;D-galactosamine,D-GalN;Acute liver failure;Inflammation;Mouse
Q95-33,R394.2
A
1005-4847(2014)03-0015-05
10.3969/j.issn.1005-4847.2014.03.003
2014-01-21
十二五传染病重大专项(2012ZX10004-502);973课题(2012CB518905)。
吴小红(1967年-),女,实验师,研究方向:实验病理学。E-mail:bwuxiaohong@aliyun.com
郭彦(1981年-),女,实验师,研究方向:病原感染致病机制研究。E-mail:muhan0425@163.com
孙世惠(1968年-),女,副研究员,E-mail:sunsh01@163.com;周育森,男,研究员,Email:yszhou@bmi.ac.cn
