第一性原理研究FeAl金属间化合物脆性
2014-05-09俞泽民皮倩倩
俞泽民,皮倩倩
(哈尔滨理工大学 材料科学与工程学院,黑龙江 哈尔滨 150040)
第一性原理研究FeAl金属间化合物脆性
俞泽民,皮倩倩
(哈尔滨理工大学 材料科学与工程学院,黑龙江 哈尔滨 150040)
为了研究FeAl金属间化合物的脆性,采用第一性原理计算方法,建立了FeAl超晶胞模型,对H原子掺杂前后的FeAl的电子结构进行计算。计算结果显示:在B2型金属间化合物FeAl中,Al原子的sp杂化轨道具有较强的方向性,参与形成的FeAl之间pd极化键导致了晶体的脆性。在H原子掺杂浓度为1.82%的情况下,FeAl体系中置入H原子,与H原子近邻的Al原子状态发生改变,更多的晶格电子为了与H原子成键,转化为共价电子,使晶体内局域金属性下降。由于H原子参与成键,晶体内形成具有明显各向异性的键络,更易于解理。
金属间化合物;FeAl;脆性;第一性原理
引言
近年来,金属间化合物被公认为是航空材料和高温结构材料领域内具有重要应用价值的新材料。尽管Fe-Al金属间化合物的研究起步较晚,但因为Fe-Al金属间化合物包含Fe、Al两种普通金属元素,不含或少含战略性合金元素(如Ni,Cr等),原料成本远低于Ti-Al,Ni-Al金属间化合物,而其又具有长程有序的特殊结构,抗硫化腐蚀性能又高于后者,预计在加热元件、热交换器管、炉内紧固件、烧结多孔气体金属过滤器等方面有广泛应用,因此近年来受到了广泛的关注[1,2]。
然而,由于Fe-Al系金属间化合物存在低温脆性和温度超过600℃后材料强度和蠕变抗力急剧下降两个致命的弱点,因此这类合金未能作为结构材料在工业界得到实际应用。FeAl和Fe都是bcc型结构,具有同样的滑移系,但是α-Fe的延伸率一般可达40%~50%,而FeAl在真空条件下也只有5.4%,在空气中则只有2.2%[3],它们的塑性相差很大。近年的研究发现,试验环境对FeAl金属间化合物的脆性有至关重要的影响。进一步研究得出,Fe-Al金属间化合物的环境脆性是由于环境中的水蒸汽与Al原子作用,产生原子氢所引起的氢脆[4,5]。目前对FeAl脆性的研究还停留在实验阶段,对FeAl脆性机理的研究还比较少。采用计算机模拟B2FeAl合金的性能,成为研究FeAl基合金的重要手段[6,7]。为了研究FeAl金属间化合物的脆性机理,本文将采用第一性原理计算方法,建立Fe27Al27和Fe27Al27H超晶胞,对FeAl电子结构进行理论研究。
1 计算模型和方法
室温下,铝含量为36.5%~50%(at%)时,构成B2有序结构的二元FeAl金属间化合物,空间群pm3m,晶格常数为0.2909nm[10]。试验发现,氢原子在α-Fe中最大溶解度原子分数比约为2,几乎不引起其晶格常数的变化。由于FeAl与α-Fe均为体心立方型结构,有相同的间隙种类和数目。所以,可以推断,氢原子溶入FeAl的量也很少,几乎不引起它们晶格常数的变化。因此本文建立3×3× 3Fe27Al27和Fe27Al27H超晶胞模型,此时H原子的含量为1.82at.%。
计算由Materials Studio中的软件CASTEP包完成,CASTEP软件是一种基于密度泛函方法的从头算量子力学程序,计算中电子与电子间相互作用的交换相关效应通过GGA的PBE计算方案进行处理,电子波函数通过平面波基矢组展开。为尽量减少平面波基矢个数,本文采用了超然赝势(ultrasoft pseudopotential)来描述离子实与价电子之间的相互作用势。平面波阶段能量为300eV,设置K点网格为4×4×9。
2 结果和讨论
2.1 FeAl的电子结构

图1 FeAl分波态密度图Fig.1 The partial density of states of FeAl supercell

图2 FeAl晶体结构模型图Fig.2 The models of FeAl crystal structure
图1为FeAl金属间化合物的电子分波态密度图(PDOS),从图中可以看出,费米能级处的态密度贡献主要来自金属原子Fe的3d电子,Al原子对费米能级处的能态密度基本没有贡献,并且在整个能量范围内贡献都相对较小。同时,Fe的3d和Al的3p分波态密度发生较强共振,说明Fe和Al的相互作用主要依靠Fe的3d电子和Al的3p电子,同时Al的3s和3p电子有杂化。
2.2 布居数分析
表1是Fe27Al27体系和Fe27Al27H体系局部各元素的 Mulliken电荷数计算结果。由于Fe27Al27H体系原子较多,而晶体的性能都是由构成晶体的原子本身的性能以及原子间相互作用决定的,而后者在晶体性能上的反映主要受近邻原子的影响,所以只考虑最近邻和次近邻键即可。所以Fe27Al27和Fe27Al27H体系布居数只选取近邻H原子的晶胞数据进行分析。晶胞截图如图2所示。以此晶胞建立坐标系,可以看出,在Fe与Al形成二元FeAl过程中,Fe原子得到0.15个电子,Al原子失去0.15个电子,Fe原子与Al原子成键属于极性键,具有较强的方向性。当置入H原子后,H原子得到0.32个电子,近邻H原子的Al原子失电子数有所升高,而相应的近邻H原子的Fe原子得电子数明显降低。

表1 H置入FeAl前后体系的Mulliken电荷计算结果Table 1 The Mulliken charges calculation results of supercell before and after addition of H in FeAl
表2是H置入FeAl前后体系的布居数,可以看出,对于FeAl而言,Fe和Al之间的布居数大于Al原子和Al原子之间、Fe原子和Fe原子之间的布居数。说明FeAl成键方向性较强。对应图2可以看出,在H掺入Fe27Al27H中后,占据间隙位置,即处于两个相邻的Al原子中间,其中H原子与Al原子之间布居数明显大于H原子与Fe原子之间的布居数,说明H主要与Al原子成键作用。与H原子最近邻的Al原子和Al原子之间、和与H原子近邻Fe原子之间的布居数都变得很小,表明它们之间的共价性均减弱,晶棱上电子贫化减弱。而Fe原子和Fe原子之间的布居数均为负值,表明它们之间成键作用变的非常小。

表2 H置换入FeAl前后体系的布居数Table 2 The population of supercell before and after the addition of H in FeAl
2.3 FeAl脆性机理分析
图3为Fe27Al27和Fe27Al127H体系的分波态密度图,通过比较可以看出Fe27Al27H体系中,费米能级以下的3p电子和3s电子向低能级方向移动,而费米能级以上的3d电子共振峰则向高能级处移动,Al原子s和p轨道杂化更明显。
图4为H原子置入前后Fe27Al27体系中近邻H原子的Al原子的局域态密度图(LDOS)。图5为H原子置入前后Fe27Al27体系中的近邻H原子的Fe原子局域态密度图(LDOS)。从图中可以看到,H原子导致附近Fe原子费米能级以下3p电子向低能级方向移动,从而导致近邻H原子的Fe原子之间的重叠布居数降低。在低能级处,H原子的1s轨道和Al原子的3p轨道共振明显,说明H原子和Al原子成键作用主要靠H原子的1s轨道电子和Al原子的3p轨道电子。H原子掺入前后Fe原子3d电子态密度曲线波峰移动不明显,主要是因为在Fe27Al27H超晶胞中,3d电子轨道存在较强的局域性,Fe原子受H原子影响较小,导致近邻Fe原子3d电子态密度峰值移动不明显。而近邻H原子的Fe原子与周围Al原子之间重叠布居数的变化,主要是因为Al原子受到H原子影响,因此它们之间重叠布居数减小,成键作用减弱。
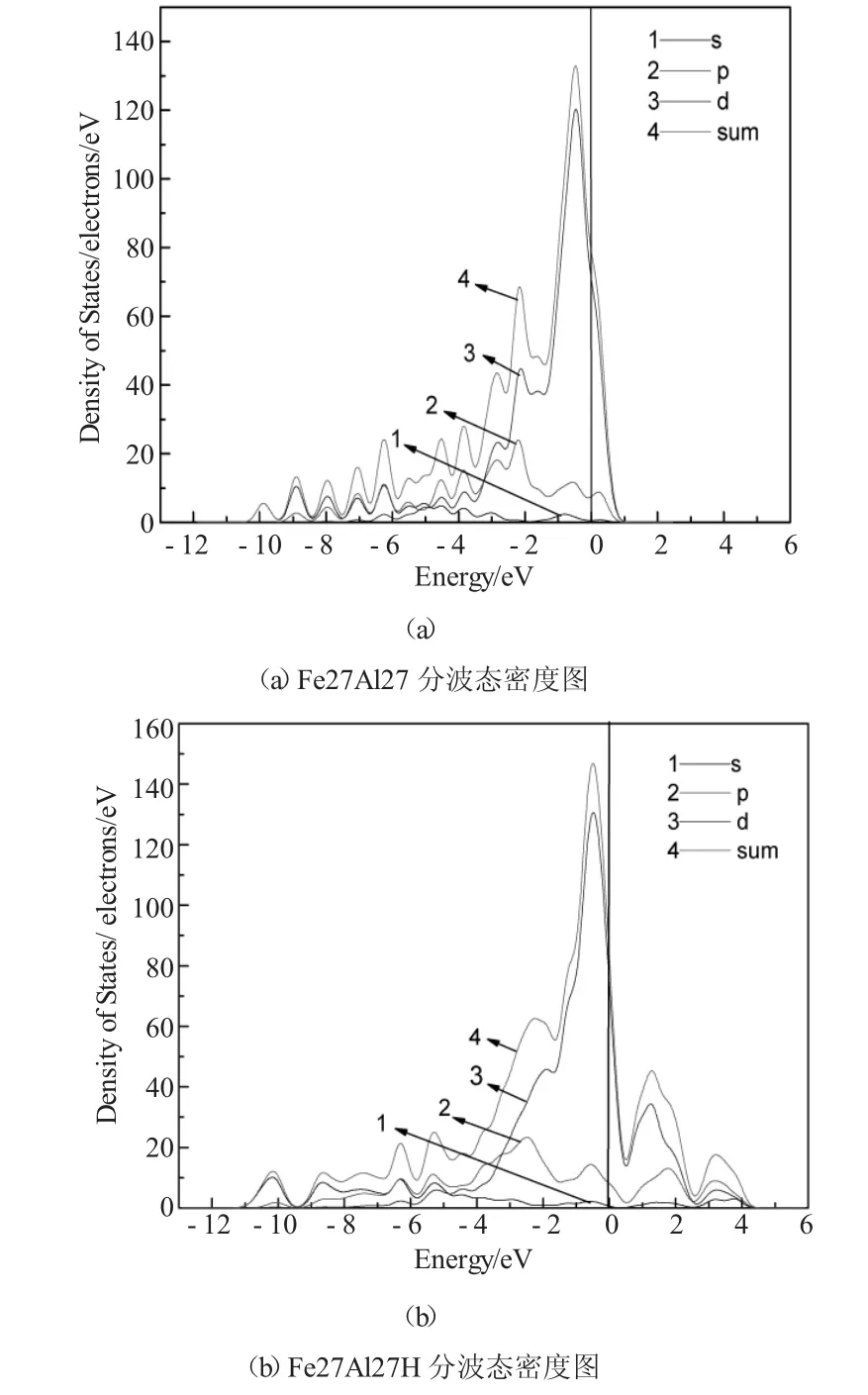
图3 Fe27Al27和Fe27Al127H体系的分波态密度图Fig.3 The partial density of states of Fe27Al27 and Fe27Al27H supercell

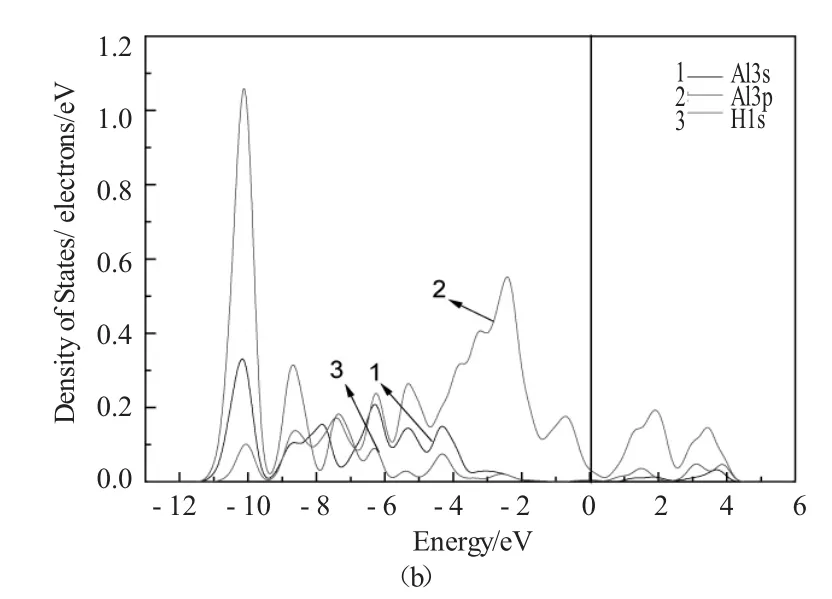
图4 Fe27Al27体系中和Fe27Al27H体系Al原子局态密度图Fig.4 The local density of states of Al atoms in Fe27Al27 and Fe27Al27H supercell
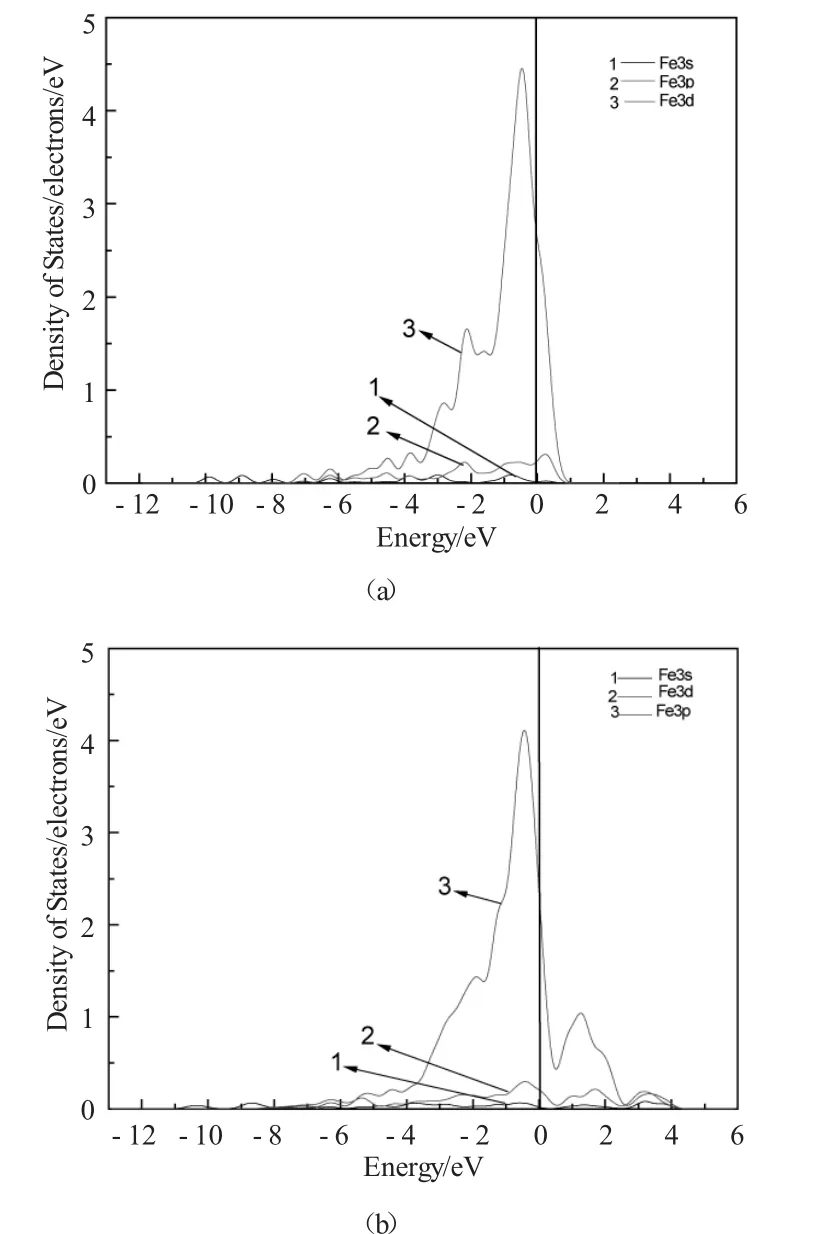
图5 Fe27Al27体系和Fe27Al27H中Fe原子局域态密度图Fig.5 The local density of states of Fe atoms in Fe27Al27 and Fe27Al27H supercell
3 结 语
经研究表明,金属间化合物的塑形与原子间成键的方向性密切相关,随着成键方向性的减弱而增大;从电子结构角度来讲,H原子的掺入对Fe的3d,Al的3p、3s电子影响,应该是H元素影响FeAl晶体电子云分布及成键方向性的主要原因。本文可得出如下结论:
1)在B2型金属间化合物FeAl中,Al原子的sp杂化轨道具有较强的方向性,参与形成的Fe-Al之间pd极化键导致了晶体的脆性。
2)在H原子掺杂浓度为1.82%的情况下,FeAl体系中置入H原子,与H原子近邻的Al原子状态发生改变,更多的晶格电子为了与H原子成键,转化为共价电子,使晶体内局域金属性下降。由于H原子参与成键,晶体内形成具有明显各向异性的键络,更易于解理。
[1]汤文明,唐红军,郑治祥,等.F e-Al金属间化合物基复合材料的研究进展[J].中国有色金属学,2003,13(4):811~825.
[2]李云峰.Fe-Al系化合物合金的制备与组织性能研究[D].兰州理工大学:2004.
[3]MEKAMEY C G,DEVANJ H,TORTORELLI P F,et al.A Review of Recent Developments in Fe3Al-Based Alloys[J]. Mater Res,1991,6(8):1779~1805.
[4]STOLOFF N S,LIU C T.Environmental embrittlement of iron alurninicles[J].Intermetallics,1994(2):75~87.
[5]林栋梁.有序金属间化合物研究的新进展[J].机械工程材料,1994,18(1):8~15.
[6]LIU C T,LEE E H,MCKAMEY C G.An environmental effects as the major cause for room-temperature embrittlement in FeAl[J].Scripta Metal Mater,1989,23(6):875~880.
[7]YOO M H,FU C L.Fundamental aspects of deformation and fracture in high-ternperature orclered intemnetallics[J].ISU Intemational,1991,31(10):1049~1062.
Study on the Brittleness of FeAl Intermetallic compounds by First Principle
YU Ze-min and PI Qian-qian
(College of Material Science and Engineering,Harbin University of Science and Technology,Harbin 150040,China)
In order to study the brittleness of the FeAl intermetallic compounds,the first-principle calculation is adopted,a supercell FeAl model is establishing for calculating the electronic structure of FeAl before and after doping H atom.The calculation results show that:in the B2 type FeAl intermetallic compounds,the sp hybrid orbital of Al atoms has a strong directivity and the pd polarization bond participating in the formation of FeAl causes the brittleness of the crystal.When the doping concentration of H atoms is 1.82%,the H atoms is put into the FeAl system and then the state of Al atom will change which is near H atoms;more lattice electrons translate into shared electron to bond with H atoms,and make the metallicity descend at part of the crystal.Since the H atoms participate in the bonding,the bond network with obvious anisotropy forms in the crystal,which makes the cleavage be more easily.
Intermetallic compounds;FeAl;brittleness;first principles
TF 64
A
1001-0017(2014)03-0196-04
2014-02-26
俞泽民(1961-)男,黑龙江人,博士,主要研究方向为金属材料强韧化、功能陶瓷与结构陶瓷、电子材料与纳米材料制备。
