一个脊髓小脑共济失调3型家系临床特征分析☆
2014-04-27李洁亮孙筱放周伯荣何文智何文茵范勇魏君朱德途
李洁亮 孙筱放 周伯荣 何文智 何文茵 范勇 魏君 朱德途
一个脊髓小脑共济失调3型家系临床特征分析☆
李洁亮*孙筱放*周伯荣△何文智*何文茵*范勇*魏君*朱德途*
目的 分析一个脊髓小脑共济失调3型家系患者临床特征,探讨脊髓小脑共济失调3型(spinocerebellar ataxia 3,SCA3)疾病家族临床异质性,为临床医生早期正确诊断疾病提供依据。方法通过聚合酶链反应及毛细管电泳片段分析确诊一个SCA3家系,详细记录患者临床特征,并对其进行汇总分析,部分患者行头颅MRI检测及眼底检查。结果此SCA3家系共18例临床患者,12例症状前患者。除经典的小脑系统症状外,其中3例还表现为智力障碍,1例伴随颈椎病,1例早期出现肌张力障碍,1例视觉系统障碍,7例植物神经机能障碍表现。部分患者MRI检测结果显示不同程度桥脑小脑萎缩,眼底检测未见明显改变。结论在同一家系中, SCA3也具有明显的临床异质性。建议当一个家系中同时存在小脑系统、视觉系统、植物神经系统障碍,颈椎病,智力障碍时,需考虑SCA3型可能。
脊髓小脑共济失调3型 临床特征 核磁共振成像 临床异质性
脊髓小脑共济失调(spinocerebellar ataxia, SCA)又称为常染色体显性遗传进行性共济失调,是小脑、脑干、脊髓系统变性而导致的以共济失调为主要表现的神经系统变性疾病[1],目前报道已经有30多种亚型[2]。脊髓小脑共济失调3型(spinocerebellar ataxia 3,SCA3),也称为Machado-Joseph病,是国内最常见的一种脊髓小脑共济失调亚型[3],目前有文献报道其所占比例超过50%[4-5]。该病的发病年龄为20~40岁,主要的临床表现有步态不稳,构音障碍,突眼,眼肌麻痹,面肌、舌肌肌束震颤,吞咽困难,饮水呛咳,锥体束及锥体外束征[6-7]。尽管目前临床已经对SCA3复杂的临床表现已经有了比较全面的认识,但是由于其临床异质性非常明显[8],有些非典型的病例仍然会被误诊。在本研究中,我们收集了一SCA3大家系,其临床表现轻重不一,表现出了多种SCA3的多种临床症状和体征,现就其临床特点进行总结,探讨和进一步拓展SCA3家族的临床异质性。
1 对象和方法
1.1 研究对象本院收集的一SCA较大家系,共48人,男23人,女25人。临床患者18例,男9例,女9例。符合常染色体显性遗传特点,诊断符合Harding标准[18]。对家系成员进行病史采集及体格检查,绘制家系图(见图1)。
1.2 头颅MRI检测家系中5人行头颅MRI检测,分别为III1,III5,III14,IV2,V1。
1.3 眼底检查接受体检成员都进行眼底检查。
1.4 ATXN3基因CAG重复数检测家系成员签署知情同意书,提取其外周血基因组DNA,运用聚合酶链反应及毛细管电泳片段分析,对所有成员进行ATXN3基因CAG重复数检测。
1.5 统计学方法本研究的资料为计数资料,采用一般描述统计方法。
2 结果
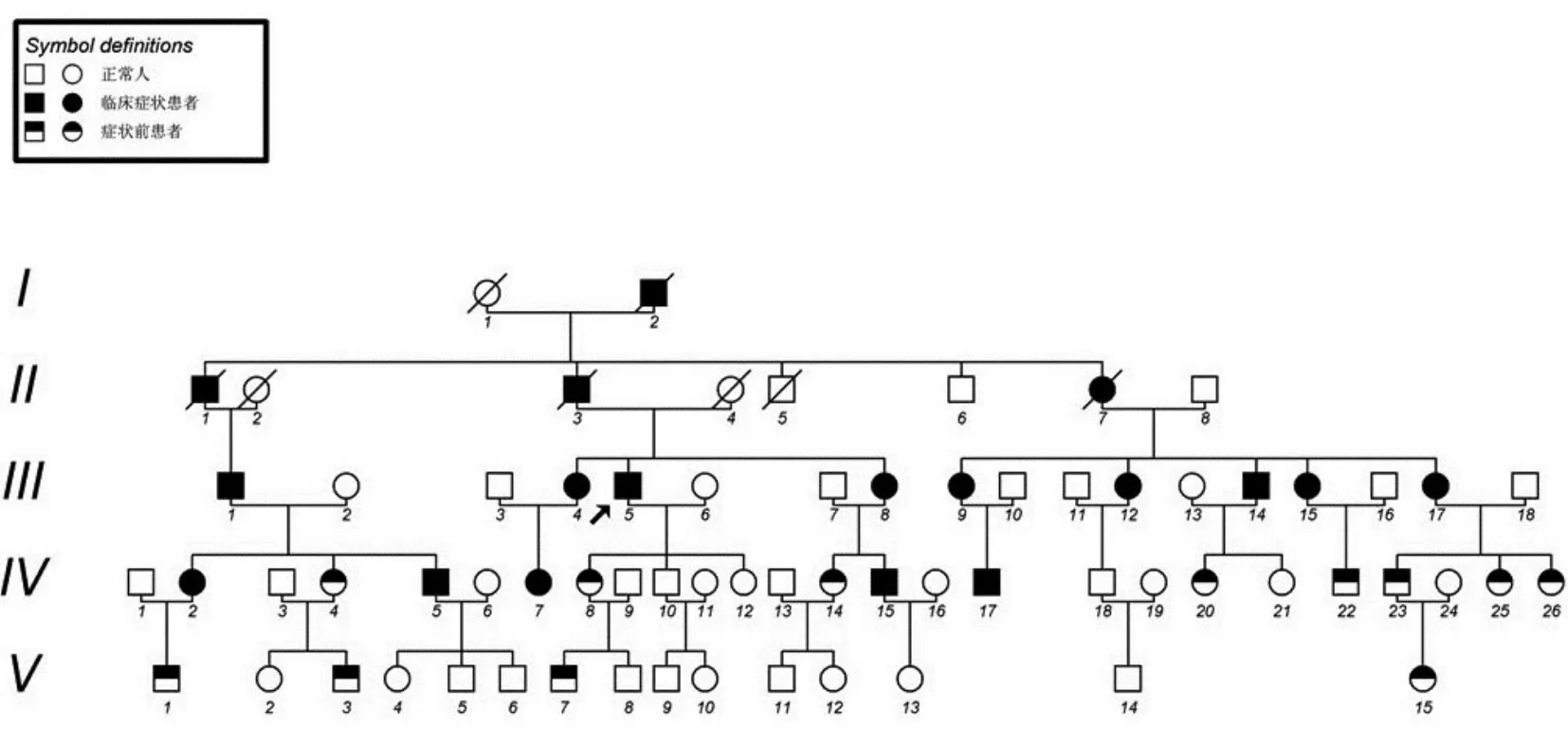
图1 脊髓小脑共济失调3型(SCA3)家系图。5代共30例患者,女16例,男14例,符合常染色体显性遗传特征。粗箭头所指为先证者
2.1 临床特征该家系中临床患者18例,男9例,女9例,符合常染色体显性遗传特点。发病年龄24~55岁,平均39.5岁;病程1~13年,平均7年。接受体格检查24名,男11名,女13名。以共济失调为首发症状者9例,以视物模糊为首发症状者1例,以双手麻木为首发症状者1例,以双膝关节疼痛为首发症状者1例。其次还表现为眼球震颤(14例),眼肌瘫痪(2例),眼突(1例),锥体束征(8例),构音障碍(3例),舌肌纤颤(5例)、萎缩(1例),浅感觉异常(6例),植物神经机能障碍(6例),颈椎病(1例)以及智力障碍(3例)。
III:1,男,58岁,“行走不稳11年,肢体麻木2年”就诊。2002年因意外左眼外伤,此后开始出现走路不稳,症状逐渐加重,目前已无法独立行走,于2011年开始出现手麻,口齿不清,伴吞咽困难、饮水呛咳,患者生活基本不能自理,病程中饮食睡眠尚可,有排汗困难,视力逐渐下降,近2年有排小便不畅感。2002年右眼外伤史,2012年行股骨头坏死手术,具体不详。查体:血压140/90 mmHg,神志尚清,智力下降,构音障碍,左眼盲,右眼视力下降,无眼球震颤,右眼瞳孔直径2.5,位置居中,对光反射灵敏。伸舌偏左,舌颤,无舌肌萎缩。肌张力增强,腱反射亢进,双侧指鼻试验、左侧跟膝胫试验、轮指快复试验阳性,双侧病理征阳性,植物神经机能障碍表现有少汗。共济失调步态,Romberg征阳性,双上肢浅感觉下降。
先证者,III:5,男,63岁,“行走不稳13年,肢体麻木1年”就诊。13年前无明显诱因出现走路不稳,呈进行性加重,6年前已无法独立行走,1年前无明显因出现肢体麻木,表现为左手麻木,伴语言不清,饮水呛咳,视物模糊。高血压病史2年,无手术外伤史。查体:血压126/80mmHg,神志清楚,智力下降,构音障碍,双侧眼球震颤,双瞳孔等大等圆,对光反射灵敏。伸舌居中,舌肌纤动,无舌肌萎缩,肌张力增强,腱反射亢进。双侧指鼻试验、跟膝胫试验、联带运动阳性。双侧病理征阳性,植物神经机能障碍表现有少汗。共济失调步态,左上肢浅感觉下降。
III:4,女,68岁,“言语不清伴行走不稳10年”就诊。患者10年前无明显诱因出现言语不清,表现为构音障碍,伴走路不稳,呈进行性加重,近1年来症状明显加重,表现为行走不能,伴吞咽呛咳。患病期间大小便尚可。精神病史多年,具体不详。查体:血压120/78mmHg,意识尚清,精神状态差,智力下降,语言不能,双侧瞳孔等大等圆,直径2.5mm,对光反射灵敏,眼突明显,眼外肌瘫痪,调节反射障碍,双侧眼球震颤。伸舌居中,舌肌纤动,舌肌萎缩存在。双侧指鼻试验、轮指快复实验、跟膝胫试验阳性。肌张力增高明显,腱反射亢进。病理征阳性,植物神经机能障碍表现为多汗。共济失调步态,Romberg's征阳性。
2.2 磁共振检测结果家系中5名行头颅MRI检测,其中4例为临床患者,均有不同程度的桥脑小脑的萎缩。1为症状前患者,尚无明显的改变,见图2(A~E)。
2.3 眼底检查该家系眼底检测大部分成员未见明显阳性改变,其中患者III:12右眼黄斑病变,患者IV:25视力差,屈光不正。见图2(F~G)。
2.4 ATXN3基因CAG重复数检测结果本次接受基因检测者为42名,男18名,女24名。经遗传学筛查后可知,该家系共26例携带突变的ATXN3基因,男11例,女15例。患者ATXN3基因异常扩增的CAG重复数为68~78,即确诊为SCA3型,见图3。
3 讨论
脊髓小脑共济失调(SCA)又称为常染色体显性遗传进行性共济失调,是小脑、脑干、脊髓系统变性而导致的以共济失调为主要表现的神经系统退行性疾病。脊髓小脑共济失调3型(SCA3)又称为Machado-Joseph病,1972年由Nakano首次报道了来自于葡萄牙Azores的一个美国家系,不同国家、地区或种族脊髓小脑共济失调发病率和不同亚型的分布存在差异。大多数国家如巴西、葡萄牙、日本、德国、荷兰等以SCA3最为常见[3]。临床主要表现为小脑性共济失调、构音障碍,眼外肌麻痹、肌痉挛强直等锥体系、锥体外系症状以及周围神经病变,病程不同阶段可见面舌肌震颤、肌萎缩、肌张力障碍、复视、突眼。感觉缺失和帕金森样表现等症状[2]。SCA3的致病基因是SCA/ ATXN3,该基因定位于14q32.12,含有11个外显子,由于第10个外显子编码谷聚酰胺的胞嘧啶-腺嘌呤-鸟嘌呤(CAG)重复序列异常扩增而导致发病的。SCA3的MRI主要表现为小脑、脑干和脊髓的不同程度萎缩[9]。遗传性共济失调具有高度的临床变异性[10],SCA3/MJD尤为显著[11],同时SCA3也具有其他SCA亚型的特征[12],因此为了准确描述其临床表型,建议将本病分成4型[13-14]。第1型,患者表现明显的张力障碍-强直综合征,发病早,共济失调少见;第2型,患者明显的小脑共济失调,中年发病;第3型,患者表现出小脑症状,周围神经病,晚期发病;第4型,患者表现为神经病症状,帕金森综合征,发病时间不一。另外,有些SCA3病人表现为痉挛性截瘫,而没有小脑共济失调症状,这被认为是第5型[15]。这些亚型可以随着疾病的进展相互转变,同一病人也可以同时表现2种以上表型,这使得SCA3的临床表现更加复杂,因此,在国内,临床医生一般也不会单一的把病人划分到固定的一种亚型。
本研究家系,经基因检测证实为SCA3型,ATXN3基因CAG重复数为68~78,平均为73次,超过了正常值范围12~44。在48位成员中,患者总数为30,其中临床患者18例,男9例,女9例;症状前患者12例,男6例,女6例,男女患病比例无差别,符合常染色体显性遗传特点,经基因检测的42人中,26例携带突变的ATXN3基因,占61.9%。该家系患者发病年龄为24~55岁,平均39.5岁,与之前的报道一致[5];病程1~13年,平均7年,症状都是进行性加重,病程超过10年的患者目前已经无法独立行走[16],需借助轮椅或家人协助。在以往的文献报道中,关于SCA3的临床异质性,很多研究者关注的都是不同家系的SCA3患者间临床表型的异质性,而在本研究家系中,我们发现在同一SCA3家系中,临床表型也存在明显的异质性。在该SCA3家系中,13例患者以共济失调为首发症状,占72.2%,为该家系的主要首发症状,与其他报道一致;除此之外,有5例分别以视物模糊、双手麻木、双膝关节疼痛、癫痫及精神病为首发,各占5.55%。其次14例患者伴随眼球震颤,为该家系突出的阳性体征;2例眼肌瘫痪,1例突眼,所以突眼在本研究家系中不典型[18];8例锥体束征,包括肌张力的下降或亢进,腱反射亢进;3例构音障碍;5例舌肌纤颤,其中1例还伴随有舌肌的萎缩;6例浅感觉异常,主要表现为肢体的麻木,浅感觉的下降;6例植物神经机能障碍,包括
多汗、少汗、大小便异常以及月经异常;3例智力障碍及1例伴有颈椎病。至于在同一家系中,对于临床异质性的原因,我们目前考虑与患者性别、SCA3/ATXN3基因CAG重复数,发病年龄等其他因素相关,相关数据进一步验证中。
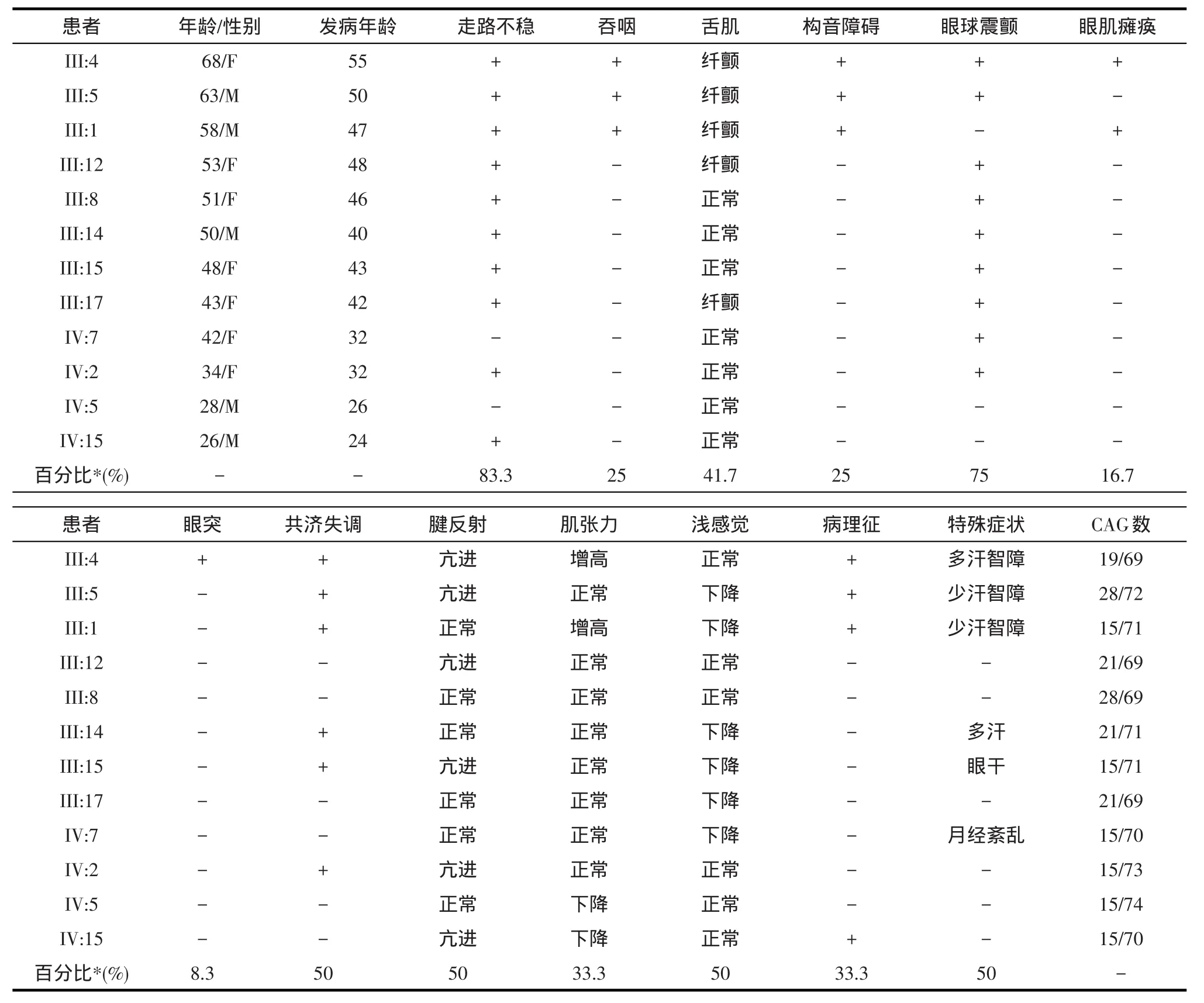
表1 家系中12例SCA3患者的临床表征


图2 头颅磁共振与眼底检查数字图像图A:III:5,桥脑小脑萎缩,双侧额顶叶、双侧侧脑室旁脱髓鞘(桥脑体积缩小,小脑中脚及小脑半球萎缩,脑沟增宽,延髓、桥脑可见线样长T2信号,脑室系统扩大)。图B:III:1,桥脑小脑萎缩,双侧额叶脱髓鞘(桥脑体积缩小,小脑中脚及小脑半球萎缩,延髓桥脑可见线样长T2信号,脑室系统扩大,以四脑室为主)。图C:III:14,桥脑小脑萎缩(桥脑体积未见明显缩小,小脑表面脑沟增宽,脑室系统未见明显改变)。图D:IV:2,桥脑小脑萎缩(桥脑体积未见明显缩小,双侧小脑半球、小脑蚓部脑沟增宽,桥脑可见线样长T2信号,中线结构居中,四脑室稍扩大)。图E:V:1,症状前患者,未见明显结构改变。图F:患者III:12右眼黄斑病变。图G:IV:25左眼视力0.3,右眼0.2,屈光不正
另外,本研究还对家系部分成员进行头颅MRI检测,发现家系中先证者1桥脑体积缩小,小脑中脚及小脑半球萎缩,小脑表面脑沟增宽,双侧额叶斑点状长T2信号,脑室系统扩大,以四脑室扩大为主;先证者2双侧额顶叶、双侧侧脑室旁可见斑点状长T2信号,桥脑体积缩小,小脑中脚及小脑半球萎缩,第四脑室系统扩大明显;患者III: 14桥脑体积未见明显缩小,小脑表面脑沟增宽,脑室系统未见明显改变;患者IV:2桥脑体积未见明显缩小,双侧小脑半球、小脑蚓部脑沟增宽,四脑室稍扩大;症状前患者V:1脑实质、脑室系统未见明显异常。由文献可知,SCA3的MRI主要表现为小脑、脑干和脊髓的不同程度萎缩[16],我们的检查结果与其符合。另外,结合临床症状可知,随着病程的发展,头颅MRI改变更加明显。

图3 家系部分成员基因片段分析图。1为III:1 SCA3/ATXN3基因的正常CAG重复数为15(黑色箭头所示),异常重复数为71(红色箭头所示);2为先证者SCA3/ATXN3基因的正常CAG重复数为28(黑色箭头所示),异常重复数为72(红色箭头所示);3为III:14,SCA3/ATXN3基因的正常CAG重复数为21(黑色箭头所示),异常重复数为71(红色箭头所示);4为III:4 SCA3/ATXN3基因的正常CAG重复数为19(黑色箭头所示),异常重复数为69(红色箭头所示)
对本研究家系患者进行眼底检查,未发现与该疾病明确相关的眼底改变。其中,患者III:12右眼黄斑病变,患者IV:25左眼视力0.3,右眼0.2,屈光不正,考虑为个体因素所致。有文献可知,SCA7型容易合并黄斑萎缩、视网膜色素变性[17],眼底检测可以很好的鉴别分型。
本研究详细的描述了一个SCA3大家系患者的临床特点,并对患者进行了全面的体格检查,在很大程度上补充了SCA3疾病患者的临床特征,更突显了SCA3疾病的临床异质性,证明临床异质性在同一家系中存在,指出SCA3病人可能伴随的少见表型如智力障碍、浅感觉下降、颈椎病、精神类疾病以及植物神经机能障碍。这些非典型的临床症状可能会使很多SCA3病人在早期得到误诊。所以我们建议,当一个SCA家系中的患者分别存在小脑系统、视觉系统、植物神经系统障碍,颈椎病,智力障碍时,需考虑SCA3型可能。
[1] Schols L,Bauer P,Schmidt T,et al.Autosomal dominant cerebellar ataxias:clinical features,genetics and pathogenesis[J]. LancetNeurol,2004,3(2):291-304.
[2] ZHAO Y,LIM SW,TAN EK.Genetic analysis of SCA27 in ataxia and childhood onset postural tremor[J].Am JMed Genet BNeuropsychiatr Genet,2007,144(3):395-396.
[3] Dürr A,Stevanin G,Cancel G,et al.Spinocerebellar ataxia 3 and Machado-Joseph disease:clinical,molecular,and neuropathological features[J].Ann Neurol,1996,39(4):490-499.
[4] Maciel P,Gaspar C,DeStefano AL,et al.Correlation between CAG repeat length and clinical features in Machado-Joseph disease[J].Am JHum Genet,1995,57(1):54-57.
[5] Gan SR,Shi SS,Wu JJ,et al.High frequency of Machdo-Joseph disease identified in southeast chinese kingdreds with spinocerebellar ataxia[J].BMCMed Genet,2010,11(1):47-55.
[6] Tang B,Liu C,Shen L,et al.Frequency of SCA1,SCA2,SCA3/ MJD,SCA6,SCA7 and DRPLA CAG trinucleotide repeat expansion in patientswith hereditary spinocerebellar ataxia from Chinese Kindreds[J].Arch Neurol,2000,57(4):540-544.
[7] Ichikawa Y,Goto J,HattoriM,et al.The genomic structure and expression of MJD,the Machado-Joseph disease gene[J].JHum Genet,2001,46(3):413-422.
[8] 顾卫红,王国相,王康,等.脊髓小脑共济失调3型临床变异型特征及突变分析[J].中国现代神经疾病杂志,2008,8(2):134-138.
[9] 贾丹丹,江泓,唐北沙.马查多-约瑟夫病的分子遗传学研究进展[J].中华遗传学杂志,2008,25(6):660-663.
[10] Maruyama H,Nakamura S,Matsuyama Z,et al.Molecular features of the CAG repeats and clinicalmanifestation of Machado-Joseph disease[J].Hum MolGenet,1995,4(8):807-812.
[11] Warrick JM.Ataxin3 suppresses polyglutamine neurodegeneration in Drosophila by a ubiquitin-associated mechanism[J]. MolCell,2005,18(1):37-48.
[12] Gu W,Ma H,Wang K,etal.The shortestexpanded allele of the MJD1 gene in a Chinese MJD kindred with autonomic dysfunction[J].Eur Neurol,2004,52(1):107-111.
[13] 唐北沙,夏家辉,王德安,等.遗传性脊髓小脑型共济失调的CAG三核苷酸突变检测[J].中华医学遗传学杂志,1999,16(2):281-284.
[14] 黄智恒,徐评议,梁秀龄.广东汉族人遗传性脊髓小脑型共济失调基因突变的研究[J].中国神经精神疾病杂志,2002, 28(3):248-251.
[15] 杨笑,范学文,王进,等.宁夏地区回、汉族脊髓小脑共济失调家系SC3/MJD基因突变研究[J].中国现代神经疾病杂志,2008,8(5):556-560.
[16] 赵文新,朱武生.橄榄脑桥小脑萎缩与十字征[J].脑与神经疾病杂志,2006,14(6):433-434.
[17] Michalik A,M artin JJ,Broeckhoven CV.Spinocerebellar ataxia type 7 associated with pigmentary retinal Dystrophy[J].Eur J Hum Genet,2004,12(1):2-15.
[18] Kawaguchi Y,Okamoto T,TaniwakiM,et al.CAG expansions in a novle gene for Machado-Joseph disease at chromosome 14q32.1[J].NatGenet,1994,8(3):221-228.
[19] 利婧,张成,詹益鑫,等.脊髓小脑共济失调3型家系CAG动态突变分析和产前诊断[J].神经系统遗传性疾病,2008,12(3):282-287.
The Analysis of Clinical Manifestations in a Large SCA 3 Pedigree.
LI Jieliang,SUN Xia of ang,ZHOU Borong, HE Wenzhi,HE Wenyin,FAN Yong,WEI Jun,ZHU Detu.
Key Lab for Major Obstetric Diseases of Guangdong Province, Experimental Department of Institute of Gynecology and Obstetrics,The Third Affiliated Hospital of Guangzhou Medical University,Guangzhou 510150,China.Tel:020-81292117.
ObjectiveTo analysis the clinicalmanifestations of a large Spinocerebellar Ataxia 3 pedigree to provide the information for the early diagnosis of Ataxia 3.MethodsSCA3/ATXN3 gene was determined by using Polymerase Chain Reaction and fragment analysis in the large pedigreemembers and patients’clinical data was collected. Five patients underwentMRI imaging and fundus examination.ResultsTherewere eighteen clinical patients and twelve ATXN3 carriers in this Pedigree.In addition to ataxia,three patients presented with intellectual disability,one with cervical spondylosis,one with dysmyotonia,one with disorder in visual system,and seven with abnormality in autonomic nervous system.The MRI revealed that pons and cerebellar atrophy in some patients inordinately.Undus examination did not reveal any obvious abnormality.ConclusionsThe symptoms of SCA3 are heterogeneous in the same pedigree.When patients present with symptoms of cerebellar system,visual system and autonomic nervous system,or cervical spondylosis and intellectual disability,SCA3 should be considered.
Spinocerebellar ataxia 3 Clinicalmanifestation Magnetic resonance imaging Clinicalheterogeneity
R744
A
2013-10-18)
(责任编辑:李立)
10.3936/j.issn.1002-0152.2014.04.003
☆国家自然科学基金项目(编号:31171229)
*广东省产科重大疾病重点实验室,广州医科大学附属第三医院妇产科研究所实验部(广州 510150)
△广州医科大学附属第三医院神经内科
