Determination of azithromycin in raw materials and pharmaceutical formulations by HPLC coupled with an evaporative light scattering detector
2014-04-20XiLiuSiruoZhngYnZhengPingHungKngliDuQingFu
,Xi Liu,Siruo Zhng,Yn Zheng,Ping Hung, Kngli Du,Qing Fu,
aSchool of Pharmacy,Xi’an Jiaotong University,Xi’an 710061,China
bDepartment of microbiology,Dalian Medical University,Dalian 116044,China
cXi’an Institute of Food and Drug Control,Xi’an 710054,China
Short Communication
Determination of azithromycin in raw materials and pharmaceutical formulations by HPLC coupled with an evaporative light scattering detector
Aiguo Zenga,Xia Liua,Siruo Zhangb,Yan Zhengc,Ping Huangc, Kangli Dua,Qiang Fua,*
aSchool of Pharmacy,Xi’an Jiaotong University,Xi’an 710061,China
bDepartment of microbiology,Dalian Medical University,Dalian 116044,China
cXi’an Institute of Food and Drug Control,Xi’an 710054,China
A R T I C L E I N F O
Article history:
Received 18 November 2013
Received in revised form
27 November 2013
Accepted 26 December 2013
Available online 5 January 2014
Azithromycin
High-performance liquid chroma
tography
Evaporative light scattering detector
Determination
A simple high-performance liquid chromatography(HPLC)method coupled with an evaporative light scattering detector(ELSD)was developed for the determination of azithromycin in raw materials and pharmaceutical formulations(injections,capsules and tablets)without any pretreatment or derivatization step.Azithromycin,degradation products and formulation ingredients were separated eff i ciently by using the mobile phase consisted of ammonium acetate(0.05 M,pH 8.0)and acetonitrile(60:40,v/v)in an isocratic mode at 0.8 ml/min f l ow rate.Parameters of ELSD were 60°C for evaporation temperature and 50 psi for pressure of carrier gas(air).A logarithmic calibration curve was obtained from 50.93 to 509.30 μg/ml(r=0.9996)for azithromycin,with the limit of detection(LOD)of 6.75 μg/ml(S/n=3)and the limit of quantif i cation of 22.50 μg/ml(S/n=10).The developed method was validated and applied with satisfactory accuracy and precision for the determination of azithromycin in raw materials and pharmaceutical formulations(recovery 99-102%,RSD <1.2%,n=3).No signif i cant difference(t-test)was found between the results of the developed HPLC-ELSD method and the HPLC-UV or microbiological method. © 2014 Shenyang Pharmaceutical University.Production and hosting by Elsevier B.V.All rights reserved.
1. Introduction
Azithromycin(AZM)is a novel macrolide antibiotic and a semisynthetic-erythromycin derivative.It has a methylsubstituted nitrogen at position 9a in the lactone ring to create a 15-membered-ring macrolide[1].AZM produces an enhanced spectrum and potency against bacteria compared with other macrolides and superior stability in acid environment.Itsmechanism issimilarto erythromycin, appearing to bind to the same receptor,50s ribosomal subunits of susceptible bacteria and suppresses protein synthesis.AZM hasgreater oral bioavailability,longer elimination half-lives and much higher tissue concentrations than erythromycin in animals and humans,which plays a leading role in the treatment or prophylaxis of several diseases such as bacterial upper and lower respiratory tract infections,urinary tract infections,skin and soft tissue infections,and sexually transmitted diseases [2,3].
A number of reports have been published regarding to thedetermination ofAZM,in which microbiological method was the general content assay method[4]with disadvantages of time-consuming,low detectability and poor precision.In order to overcome these problems, several high-performance liquid chromatography(HPLC) methods have been developed.The off i cial method[5-8]for the assay of AZM in pharmaceuticals is HPLC with UV detector,but AZM has only a weak UV absorbance in the wavelength range of less than 220 nm,leading to an asymmetric peak prof i le and low column eff i ciency.Other methods,including electrochemical detection[9-12],f l uorescence detection by pre-column derivatization[13-15] and liquid chromatography-mass spectrometry(LC-MS) [16-18]have been used to determine AZM in routine pharmaceutical dosage forms or biological matrices.Obviously,electrochemical detectors are not widely available in many laboratories.The USP method[19]describes a high pH mobile phase(pH11)as well as a specif i c column“Gamma-alumina”which is quite expensive and diff i cult to obtain in order to assay AZM using an amperometric electrochemical detector.Moreover,pre-column derivatization is time-consuming due to the complex steps involved.Also, it would be unrealistic to use LC-MS for the routine quality control of AZM preparations.Therefore,it is necessary to develop a convenient and effective method for the quality control of AZM by using conventional materials,reagents and equipment.
Evaporative light scattering detector(ELSD)is described as a quasi-universal detection mode suitable for non-absorbing analytes[20-22]and end-absorbing analytes[23-25].The response does not depend on the solute optical properties,but on the size,shape and surface properties of the particle formed,any compound having lower volatility than the mobile phase can be detected.ELSD operation principle mainly consists of three successive processes:(a)nebulization of chromatographic eluent using nitrogen or air,(b)evaporation of mobile phase at relatively low temperature and(c)light scattering by the non-volatile residual particles,which ideally consist of analyte molecules[26,27].This complex mechanism leads to a non-linear empirical quantitative law described by the relation:
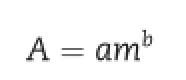
where A is the area of the chromatographic peak,m the mass of the analyte,a the response factor and b is the response index measured from the slope of the curve log A=f(logm)[i.e.logA=blogm+loga].The coeff i cients a and b depend on many parameters,such as the average size,the shape and the distribution of the particles,the nature,the volatility and the concentration of the analyte,the nature of the mobile phase and nebulizing gas,the liquid and gas f l owrates,evaporation temperature,etc.In the f i eld of pharmaceutical analysis,it has already been proposed as an effective alternative for both the determination of the aminoglycoside antibiotics,natural medicines[28],compound medicines and assessing the separation of drug combination [29].
The purpose of this study was to develop and validate a rapid and simple HPLC method with ELSD detection for the determination of AZM.The composition and f l ow rate of mobile phase and the column temperature of the method were optimized by using an orthogonal experimental design at four levels.The results proved the established method is precise,accurate,robust and practical,and is suitable for the routine quality control of AZM in raw materials and pharmaceutical dosage forms.
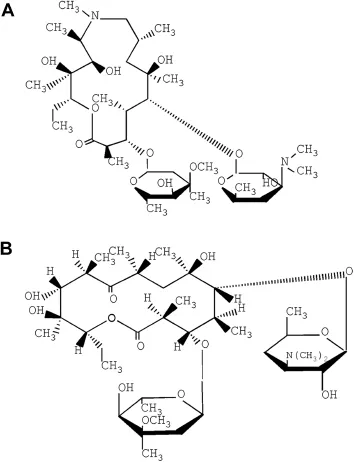
Fig.1-The chemical structures of AZM(A)and erythromycin(B).

Table 1-L16(45)5 factors-4 levels orthogonal test.
2. Materials and methods
2.1. Materials
HPLC grade acetonitrile was obtained from Tianjin Concord InternationalTrade Co.,Ltd.(China).Analyticalgrade ammonium acetate was supplied by Tianjin Kermel Chemical Reagent Co.,Ltd.(China).Other chemicals were of analytical grade.Waterwas purif i ed by redistillation and passed through a 0.45 μm membrane f i lter before use.AZM standard(931 units per mg),AZM reference substance(92.2%of purity)and erythromycin standard(931 units per mg)were purchased from the National Institute for Control of Pharmaceutical and Biological Products(Beijing,China).The structures of these compounds are shown in Fig.1.The raw materials and formulations(injections,capsules,tablets)of AZM were provided by local pharmaceutical companies (Xi’an, China), accompanied with certif i cate of analysis based on Chinese Pharmacopoeia.
2.2. Apparatus and chromatographic conditions
Liquid chromatography was performed with Agilent 1200 system(Agilent Technologies,USA)equipment comprising a quaternary pump,a G1329 A autosampler and a G1316 A column oven.Chromatographic separation was performed on 250 mm × 4.6 mm,i.d.,5 μm particle size,pore diameter 100 A°,Boston pHlex ODS reversed phase column at 40°C with the mobile phase consisted of ammonium acetate(0.05 M,pH 8.0) and acetonitrile in the proportion of 60:40(v/v).Before being mixed with the acetonitrile,the ammonium acetate(0.05 M) was adjusted to pH 8.0 by triethylamine and f i ltered through 0.45 μm nylon membrane.The f l ow rate of the mobile phase was 0.8 ml/min and the volume of each injection was 10 μl.The detector used was a Model 300s evaporative light scattering detector(SofTA Corporation,USA).The drift tube temperature of the ELSD was 60°C.The nebulizer gas was air of industrial purity grade provided by a CA-2 quiet oil-free air pump and the pressure of nebulizing gas was 50 psi.In these conditions AZM retention time was roughly 16 min.All data acquired were processed by Agilent Chemstation Rev.B.04.01 software(Agilent,Palo Alto,CA).
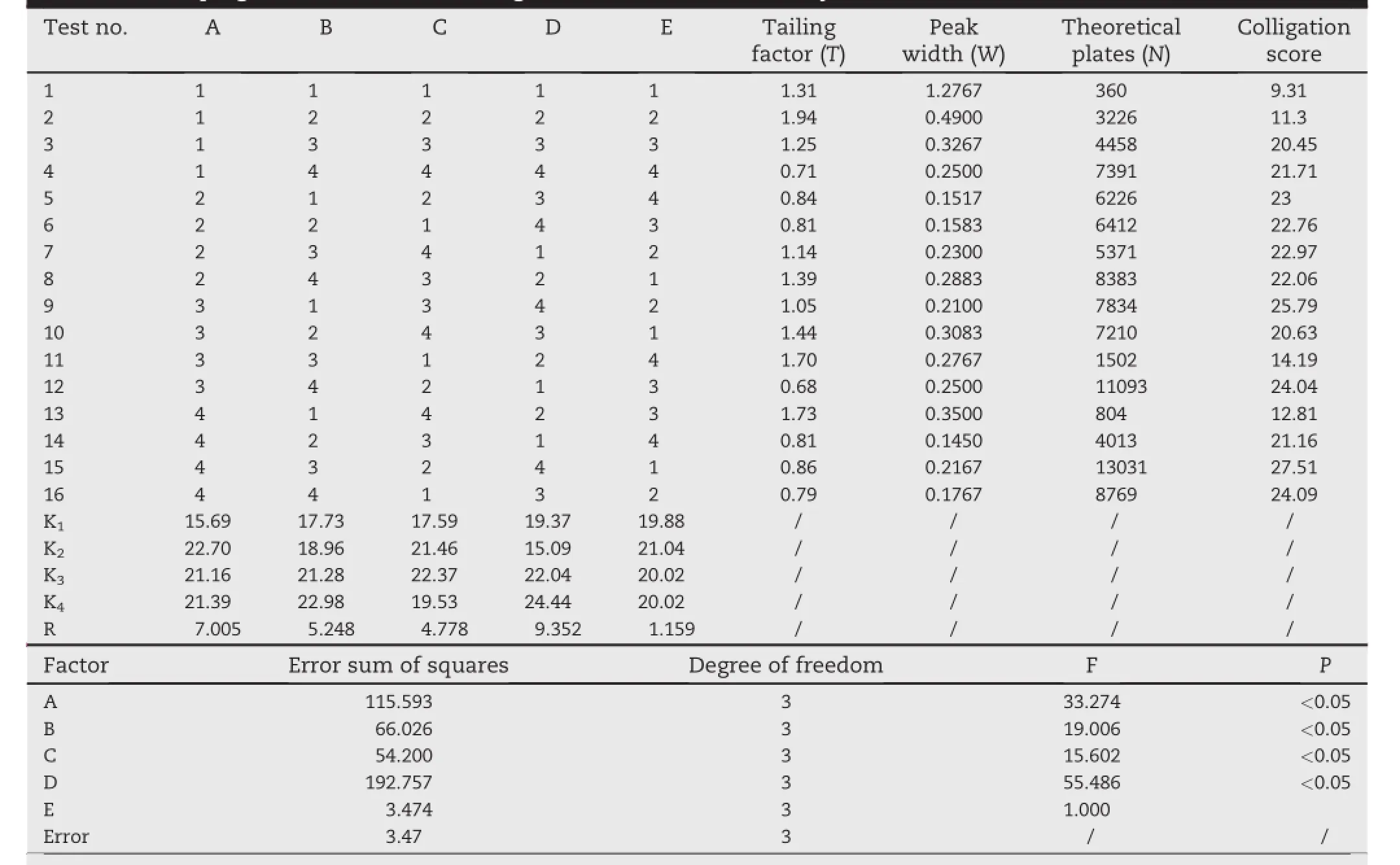
Table 2-The program and results of orthogonal test and variance analysis.
2.3. Solution preparation
2.3.1. Stock and working standard solutions
StocksolutionsofAZMwereprepared,55.24mgAZMreference substance(equivalent to 50.93 mg AZM)was transferred to a 100mlvolumetricf l ask,followedbytheadditionofabout50ml mobilephaseandsonicatedfor15min,thenmadeupto100ml withmobilephase.Workingstandardsolutionswereprepared by further dilution of the AZM stock solution with the mobile phasetofurnishf i vedifferentconcentrations withintherange of interest,in this work 50.93,101.86,152.79,254.66 and 509.30 μg/ml,then f i ltered through a 0.45 μm membrane f i lter.
2.3.2. Reference standard solution
tandard solution of AZM was prepared at concentration of 0.25 mg/ml,and used for the evaluation of the precision and stability of the method.
2.4. Sample preparation
Drug substances were accurately weighed and dissolved in mobile phase to obtain a concentration level within the working range.Concentrations of AZM solution were 0.25 mg/ ml for the assay of AZM.
2.5. Procedure
Before measurements,the column was equilibrated for at least 1 h with mobile phase f l owing through the chromatographic system until baseline noise became negligible (less than 0.5 mV at detector gain 8).10 μl of the standard or sample solutions were injected into the chromatograph using conditions described above(each solution was injected in twice).
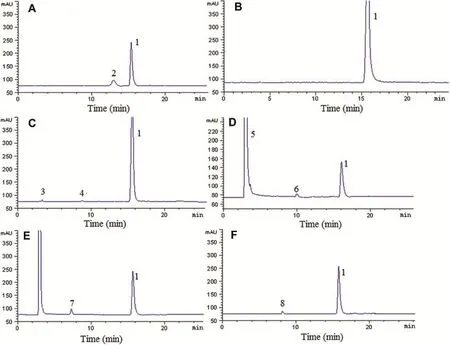
Fig.2-Representative chromatograms of erythromycin and AZM as standard(A)and AZM raw materials exposed to natural light and UV lamp(B),and treated with heat(C),1 M alkali(D),1 M acid(E)and 1%hydrogen peroxide(F).1,AZM;2, erythromycin;3,4,5,6,7,8,degradation products(unknown impurities).
3. Results and discussion
3.1. Optimization of separation conditions
AZM contains two nitrogen atoms,which make it a weakly basic compound.There have always been problems in analyzing drugs like AZM because of their basic properties. These compounds will interact strongly with the polar ends of the HPLC column packing materials(for example,residual silanol groups),becoming adsorbed to the column and are not easily eluted by the mobile phase.Therefore,asymmetric peaks are often observed under these conditions.Moreover, ELSD demands the evaporation of the mobile phase prior to light scattering step,mobile phases of high volatility are required.Several chromatography systems were investigated by us to resolve these diff i culties:acetonitrile was selected for the organic phase instead of methanol because of its higher eluting ability.The aqueous phase,involving ammonium formate or ammonium acetate solutions,was mixed with acetonitrile.Different pH values of the aqueous phase were investigated because increasing or decreasing the ionization of AZM by pH adjustment can change the column retention of AZM.Triethylamine,an organic modif i er,was used for improving the peak shape of AZM with amino groups and increasing the baseline noise.
After a preliminary crude optimization,orthogonal experiment[30,31]was applied for the f i nal optimization of mobile phase ratio,concentration and pH values of the aqueous phase,f l ow rate of the mobile phase and the column temperature(totally f i ve parameters).Three response variables were considered for the evaluation of the eff i ciency of the chromatographic determination:(i)asymmetry factor,(ii) peak width,(iii)numbers of theoretical plate.The orthogonal design table L16(45)was used and test program is shown in Table 1 and 2.
The program and results of orthogonal test and variance analysiswere shownin Table 2,and the optimumresultswere obtained as follow:the mobile phase consisting of ammonium acetate(0.05 M,pH 8.0)and acetonitrile in the proportion of 60:40(v/v),an isocratic mode at a rate of 0.8 ml/min.The chromatogram system showed sharp symmetrical peaks and good separation,AZM and its impurities could be eluted with baseline separation in 25 min.
3.2. Optimization of ELSD conditions
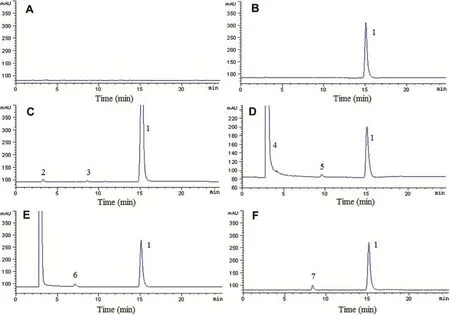
Fig.3-Representative chromatograms of AZM capsules blank excipient(A)and AZM capsules exposed to natural light and UV lamp(B),and treated with heat(C),1 M alkali(D),1 M acid(E)and 1%hydrogen peroxide(F).1,AZM;2,3,4,5,6,7, degradation products(unknown impurities).
The critical parameters of ELSD are the temperature of the tube(Ttub)and the gas pressure(Pressure).These were not included in the orthogonal experimental optimization,since they appear a minor inf l uence on the chromatographic separation comparing to mobile phase composition.However,depending on the nature of the analyte,they may appear a greater impact on the detectability and sensitivity of the method.Using the optimized mobile phase,a two-step univariate optimization was conducted.In the f i rst step,the drift tube temperature recommended was 60°C.Based on this, various temperatures range from 45 to 80°C were tested to study the inf l uence on ELSD response and signal-to-noise ratio.The results showed that the optimal parameter was identif i ed as 60°C.In the second step,using the optimum drift tube temperature,the pressure of nebulizing gas recommended was 50 psi.Based on this,various pressures(45,50 and 55 psi)were tested to study the inf l uence on ELSD response and signal-to-noise ratio,and the results showed that it is no considerable inf l uence on the detector response (peak area)was observed.Therefore,best results were obtained for 60°C and 50 psi,respectively.
3.3. System suitability
Fig.2A illustrates a typical chromatogram of AZM and erythromycin,which might be the major impurity in the synthesis of AZM[7],using the optimum chromatographic conditions. The retention time of erythromycin and AZM were 13 and 15.5 min,respectively.Two chromatographic peaks were obtained with good resolution(R=3.15)showing that AZM peak was free of interference of erythromycin.
3.4. Validation of the method
The procedures and method characteristics used for validation of this method were those described in the International Conference of Harmonization(ICH)Guidelines[32].Specif i city,linearity,precision,stability and durability of the method were evaluated.Limits of detection and quantif i cation,accuracy and repeatability were also calculated.
3.4.1. Specif i city
The ability of the chromatographic system to resolve AZM from its degradation products and formulation excipients was investigated.The raw materials,different formulations of AZM and all excipients were stored under relevant stress conditions(light,heat,acid/base hydrolysis and oxidation, respectively).Figs.2-5 show the chromatograms obtained from these samples.The samples showed light stability while degradation products were produced under heat,acid/base hydrolysis and oxidation conditions.Specif i city was demonstrated showing that neither formulation excipients nor degradation products interfered with quantif i cation ofazithromycin indicating that the proposed method can also be used in a stability assay.
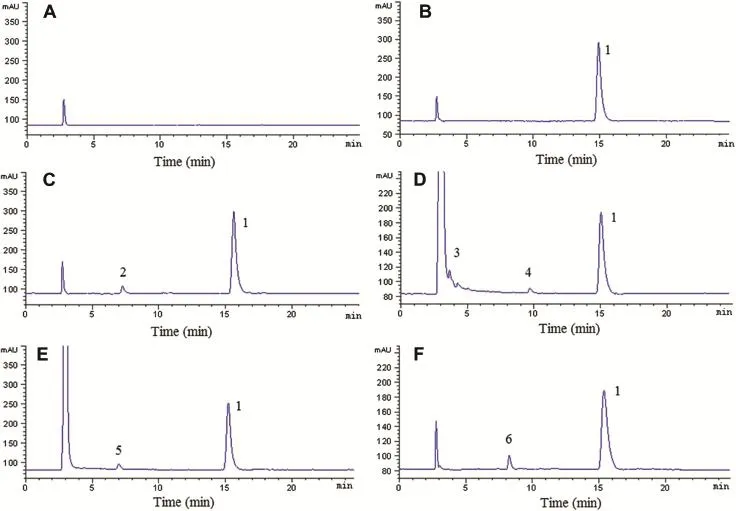
Fig.4-Representative chromatograms of AZM injections blank excipient(A)and AZM injections exposed to natural light and UV lamp(B),and treated with heat(C),1 M alkali(D),1 M acid(E)and 30%hydrogen peroxide(F).1,AZM;2,3,4,5,6, degradation products(unknown impurities).
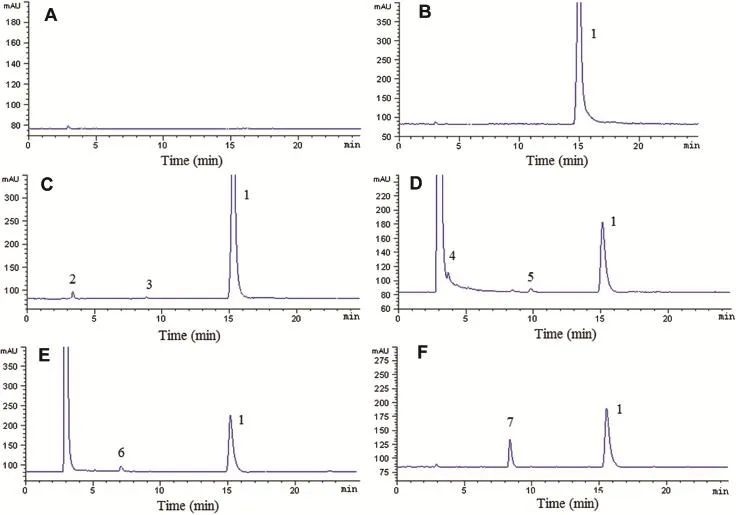
Fig.5-Representative chromatograms of AZM tablets blank excipient(A)and AZM tablets exposed to natural light and UV lamp(B),and treated with heat(C),1 M alkali(D),1 M acid(E)and 1%hydrogen peroxide(F).1,AZM;2,3,4,5,6,7,degradation products(unknown impurities).
3.4.2. Linearity of response
Linearityisdeterminedexperimentallybyanalysisofaseriesof standards at f i ve different concentrations that span at least 80-120%oftheexpectedworkingrangeoftheassay.Ithasbeen generally observed that the detector response,as measured by peak area,varies exponentially with the mass of analyte,and this behavior can be mathematically expressed in logarithmic form.Present experimental results showed that the logarithm of peak area of each standard was linearly correlated to the logarithm of injected concentration within a particular range. In this investigation,a linear plot was obtained from f i ve AZMstandardsolutionsofdifferentconcentrationintherange 50.93-509.30 μg/ml(20-200%of theoretical value)using two replicate injections.The corresponding coeff i cient(r=0.9996) was achieved with the well-established exponential relationship between peak area and sample concentration: logA=1.3142logC+0.2359.Theresultindicatedgoodlinearity.
3.4.3. Precision of the assay
The precision of the method was determined by injecting the working standard six times in succession,and the relative standard deviation(RSD)of the six peak areas was calculated. Reference standard solution and samples solution of AZM at 100%ofthe test concentration(0.25mg/ml)werepreparedand then assayed for AZM using the recommended HPLC system. RSD values were better than 1.5%.
3.4.4. Limit of detection(LOD)and quantif i cation(LOQ)
The limit of detection(LOD)is def i ned as the lowest absolute concentration of the analyte that can be accurately detected but not necessarily quantif i ed under the stated experimental condition and the limit of quantif i cation(LOQ),taken as the lowest absolute concentration of the analyte in the sample which can be determined with acceptable precision and accuracy under the stated experimental condition.Their determination both could be made by the calculation of the signalto-noise ratio.A ratio of 3 was selected and successive dilutions of the reference standard solution gave a LOD relative to the AZM peak,the LOD was 6.75 μg/ml.A ratio of 10 was selected and successive dilutions of the reference standard solution gave a LOQ relative to the AZM peak,the LOQ was 22.50 μg/ml,indicating that the HPLC-ELSD method is precise and sensitive for the quantitative evaluation of AZM.
3.4.5. Accuracy
The accuracy of the new method was evaluated by analyzing the quality control of samples spiked with standard solutionsand expressed as a percentage error of measured concentrations versus nominal concentrations(namely,recovery experiments).A recovery study was performed by analysis of simulated formulations at three concentrations,namely 80%, 100%and 120%of the labeled amount of AZM raw materials and formulations.The results of the recovery of the three concentrations of simulated AZM raw materials and formulations are summarized in Table 3.As it is clear from Table 3, all the values of RSD for different formulations recovery test were found to be within specif i ed limits,showing that the recovery of the method was satisfactory.
3.4.6. Stability
The stability of the reference and sample solutions was tested byusingthesameworkingstandard for0,1,2,4,6 and8 hwith the same mobile phase.Between runs,solutions were stored at room temperature.The areas of the f i ve peaks were calculated with RSD,RSD values were better than 2.0%.
3.4.7. Repeatability
The method repeatability was determined using six determinations at 100%of the test concentration(0.25 mg/ml), the following results are shown in Table 4.RSD values are givenfor each AZM.In everycase,RSD values werebetter than 1.5%indicating the repeatability of the proposed method is acceptable.
3.4.8. Durability
We determined the effect of different ratio of organic phase and pH values of aqueous phase,of column temperature,and of f l ow rate,using sample solution for the durability study. The chromatographic conditions investigated are listed in Table 5.It is apparent from this table that changing the column temperature had a noticeable effect on AZM chromatographic peak shape and AZM content.While,changing the fl ow rate had an obvious effect on the content of AZM but almost no effect on AZM chromatographic peak shape. Changing the ratio of organic phase and the pH of the aqueous phase had no detectable effect on the chromatographic system.
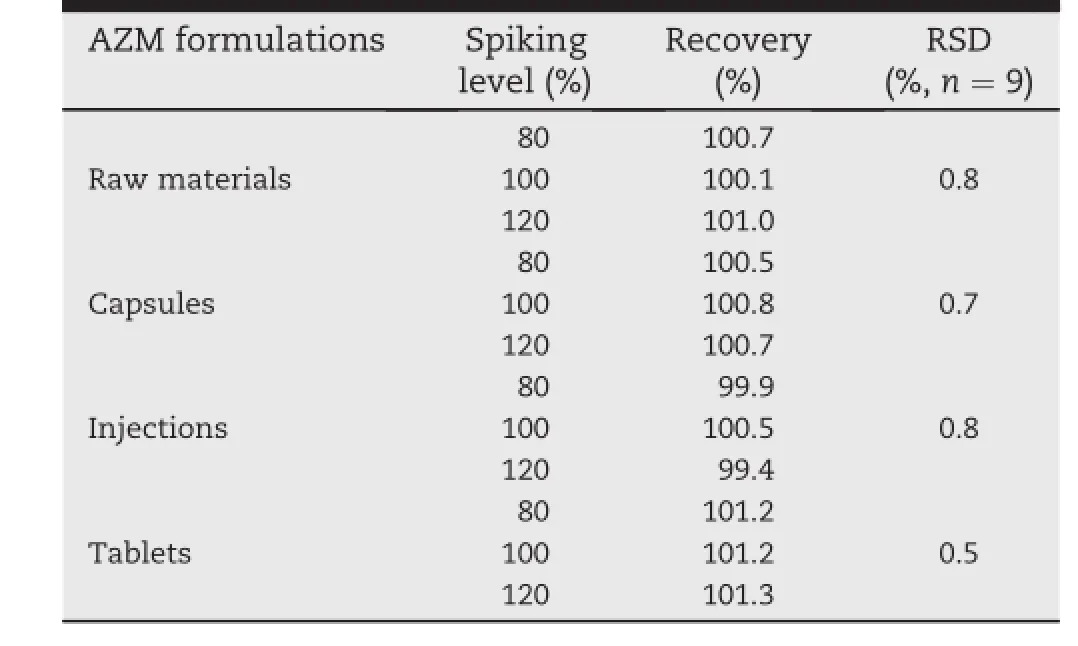
Table 3-Results of recovery test for AZM in pharmaceutical raw materials and formulations by HPLC-ELSD.
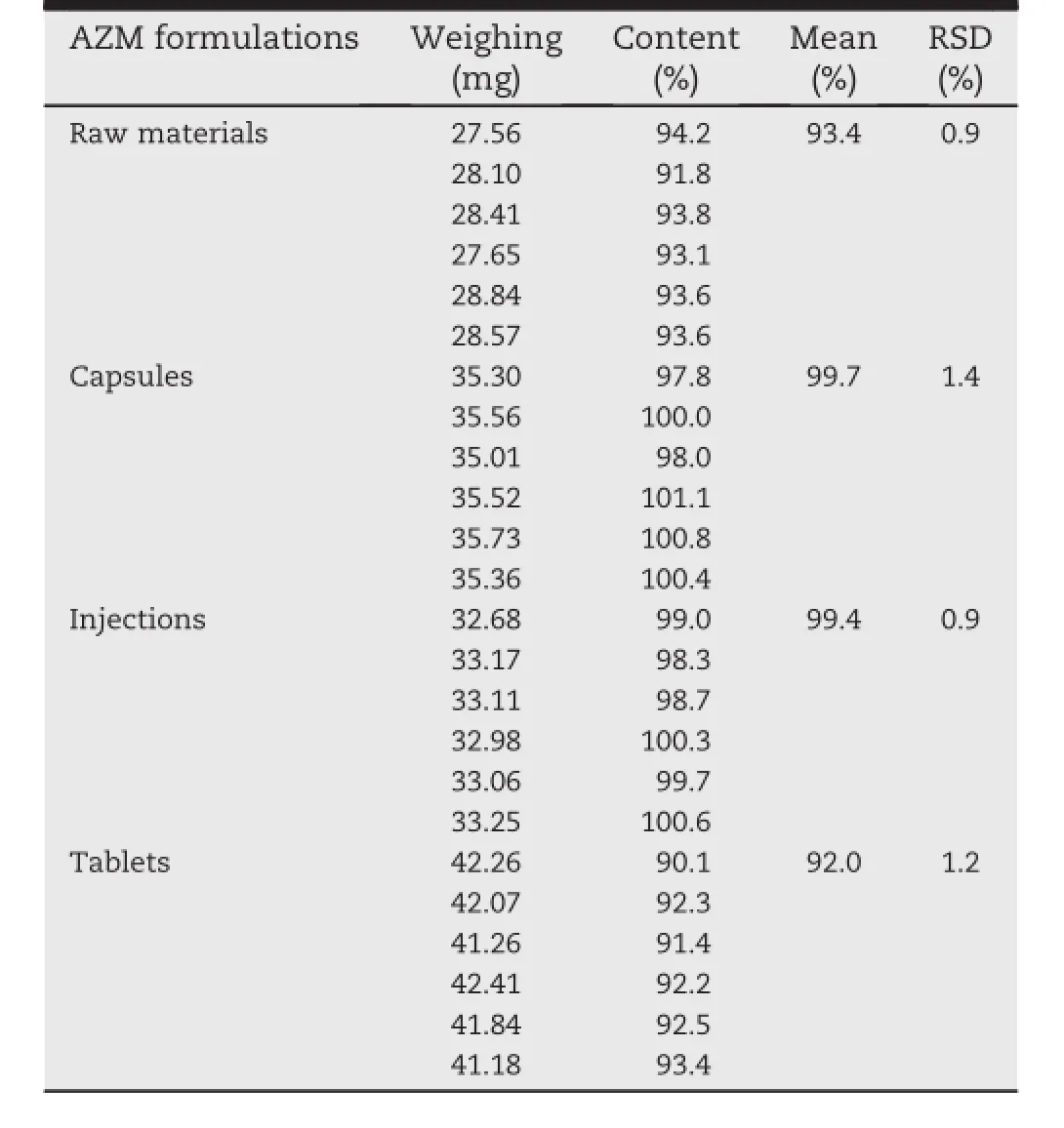
Table 4-Results of repeatability test for AZM in pharmaceutical raw materials and formulations by HPLC-ELSD.
3.5. Application to analysis of pharmaceutical formulations
The proposed HPLC-ELSD method was applied for the determinationofAZM incommercialformulationsby assaying raw materials,capsules,injections and tablets.The assay showed the AZM content of these products to be within pharmacopeial limits.The results obtained with the proposed method for the analysis of AZM were compared with the experimentaldata investigated by those standard methods(HPLC-UV[7]or microbiological method[8]).Table 6 shows the results of each method.According to the variance ratio test(F-test and t-test),the calculated values of F and t listed in Table 6 indicate that there is no signif i cant difference between the HPLC-ELSD method and HPLC-UV or microbiological method.
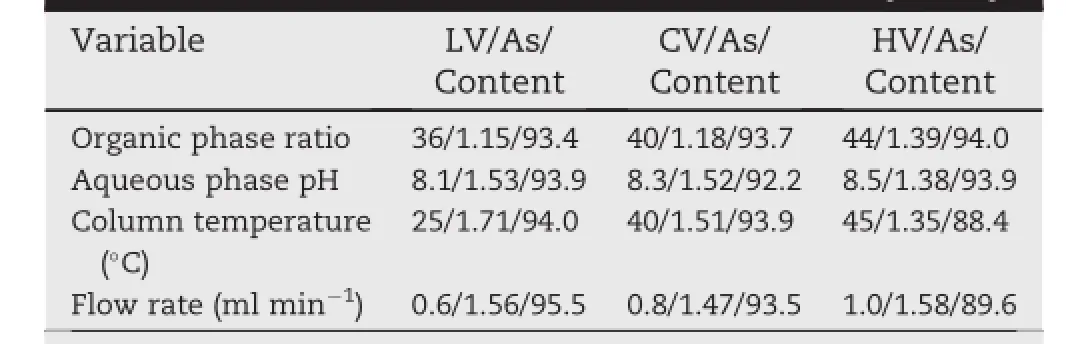
Table 5-Values of variables used in the durability study.
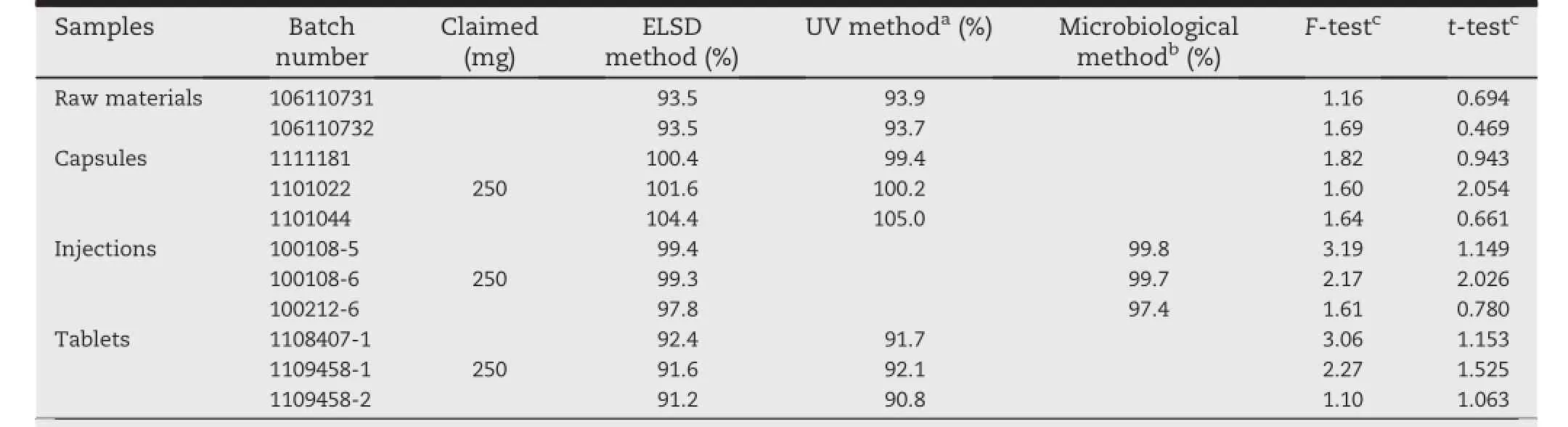
Table 6-Assay of AZM antibiotic in pharmaceutical raw materials and formulations with the ELSD and UV or microbiological method(n=4).
4. Conclusion
As is shown by the method validation and method comparison,therewasno signif i cantdifferencebetween the HPLC-ELSD method and the standard methods(HPLC-UV or microbiological method).Despites the logarithmic relationship of ELSD signal to the analyte concentration,strong detectability,precision,accuracy and repeatability were obtained.Therefore,a simple high-performance liquid chromatography(HPLC)method coupled with an evaporative light scattering detector(ELSD)appears to be eff i cient for the qualif i cation and quantif i cation of AZM in raw materials and pharmaceuticalpreparations(capsules,tabletsand injections) without any pretreatment or derivatization step.
Acknowledgments
Financial support of this work by National Natural Science Foundations of China(No.81173024)to professor Qiang Fu is gratefully acknowledged.The authors also thank Xi’an Institute of Food and Drug Control for providing the facilities and necessary equipments.
R E F E R E N C E S
[1]Fouda H,Schneider R.Quantitative determination of the antibiotic azithromycin in human serum by high performance liquid chromatography(HPLC)-atmospheric pressure chemical ionization mass spectrometry:correlation with a standard HPLC electrochemical method.Ther Drug Monit 1995;17(2):179.
[2]Christopher J,Barradel L.Azithromycin:a review of its pharmacological properties and use as three-day therapy in respiratory tract infection.Drugs 1996;51:483-505.
[3]Foulds G,Shepard R,Johnson R.The pharmacokinetics of azithromycin in human serum and tissues.J Antimicrob Chemother 1990;25:73-82.
[4]Breier A,Garcia C,Oppe T.Microbiological assay for azithromycin in pharmaceutical formulations.J Pharm Biomed Anal 2002;29:957-961.
[5]Zubata P,Ceresole R,Rosasco M,et al.A new HPLC method for azithromycin quantitation.J Pharm Biomed Anal 2002;27:833-836.
[6]Yang Z,Wang L,Tang X.Determination of azithromycin by ion-pair HPLC with UV detection.J Pharm Biomed Anal 2009;49:811-815.
[7]National Pharmacopoeia Committee.Chinese pharmacopoeia,vol.2;2010.
[8]Salgado H,Roncari A.Microbiological assay for the determination of azithromycin in ophthalmic solutions.Acta Pharmaceutica Sinica 2005;40:544-549.
[9]Gandhi R,Kaul C,Panchagnula R.Validated LC method for in-vitro analysis of azithromycin using electrochemical detection.J Pharm Biomed Anal 2000;23:1073-1079.
[10]Nigovic B,Simunic B.Voltammetric assay of azithromycin in pharmaceutical dosage forms.J Pharm Biomed Anal 2003;32:197-202.
[11]Palomeque M,Ortı´z P.New automatized method with amperometric detection for the determination of azithromycin.Talanta 2007;72:101-105.
[12]Farghaly O,Mohamed N.Voltammetric determination of azithromycin at the carbon paste electrode.Talanta 2004;62:531-538.
[13]Bahrami G,Mirzaeei S,Mohammadi B.High performance liquid chromatographic determination of azithromycin in serum using f l uorescence detection and its application in human pharmacokinetic studies.J Chromatogr B Analyt Technol Biomed Life Sci 2005;820(2):277-281.
[14]Wilms E,Trumpie H,Veenendaal W,et al.Quantitative determination of azithromycin in plasma,blood and isolated neutrophils by liquid chromatography using pre-column derivatization with 9-f l uorenylmethyloxycarbonylchloride and f l uorescence detection.J Chromatogr B 2005;814:37-42.
[15]Bahrami G,Mohammadi B.A new on-line,in-tube precolumn derivatization technique for high performance liquid chromatographic determination of azithromycin in human serum.J Chromatogr B 2006;830:355-358.
[16]Chen B,Liang Y,Chen X,et al.Quantitative determination of azithromycin in human plasma by liquid chromatography-mass spectrometry and its application in a bioequivalence study.J Pharm Biomed Anal 2006;42:480-487.
[17]Chen L,Qin F,Ma Y,et al.Quantitative determination of azithromycin in human plasma by ultra performance liquidchromatography-electrospray ionization mass spectrometry and its application in a pharmacokinetic study.J Chromatogr B 2007;855:255-261.
[18]Nirogi R,Kandikere V,Shukla M,et al.Sensitive and selective liquid chromatography tandem mass spectrometry method for the quantif i cation of azithromycin in human plasma. Anal Chim Acta 2005;553:1-8.
[19]United State Pharmacopoeial Convention.United States pharmacopoeia/National formulary[S].29th ed.;2010. pp.1965-1971.Rockville,MD.
[20]Wang J,Hu X,Tu Y.Determination of spectinomycin hydrochloride and its related substances by HPLC-ELSD and HPLC-MS.J Chromatogr B 2006;834:178-182.
[21]Megoulas N,Koupparis M.Development and validation of a novel LC/ELSD method for the quantitation of gentamicin sulfate components in pharmaceuticals.J Pharm Biomed Anal 2004;36:73-79.
[22]Sarri A,Megoulas N,Koupparis M.Development of a novel method based on liquid chromatography-evaporative light scattering detection for the direct determination of streptomycin and dihydrostreptomycin in raw materials, pharmaceutical formulations,culture media and phasma.J Chromatogr A 2006;1122:275-278.
[23]Arndt J,Macko T,Bru¨ll R.Application of the evaporative light scattering detector to analytical problems in polymer science.J Chromatogr A 2013;1310:1-14.
[24]Zhou J,Hu X,Wang T,et al.HPLC analysis of egg yolk phosphatidylcholine by evaporative light scattering detector. Chinese J Chem Eng 2012;20:665-672.
[25]Narva´ez-Rivas M,Gallardo E,Rı´os J,et al.A new highperformance liquid chromatographic method with evaporative light scattering detector for the analysis of phospholipids.Application to Iberian pig subcutaneous fat.J Chromatogr A 2011;1218:3453-3458.
[26]Megoulas N,Koupparis M.Direct determination of kanamycin in raw materials,veterinary formulation and culture media using a novel Liquid Chromatography-Evaporative Light Scattering method.Anal Chim Acta 2005;547:64-72.
[27]Dreux M,Lafosse M,Morin-Allory L.The evaporative light scattering detector-a universal instrument for non volatile solutes on LC and SFC.LCGC Int 1996;9:148-156.
[28]Cong Y,Zhou Y,Chen J.Alkaloid prof i ling of crude and processed Veratrum nigrum L.through simultaneous determination of ten steroidal alkaloids by HPLC-ELSD.J Pharm Biomed Anal 2008;48:573-578.
[29]Gaudin K,Millet P,Fawaz F,et al.Investigation of porous graphitic carbon at high-temperature liquid chromatography with evaporative light scattering detection for the analysis of the drug combination artesunate-azithromycin for the treatment of severe malaria.J Chromatogr A 2010;1217:75-81.
[30]Zhang D,Chen K,Wu L,et al.Synthesis and characterization of PVA-HA-Silk composite hydrogel by orthogonal experiment.J Bionic Eng 2012;9:234-242.
[31]Franek L,Jiang X.Orthogonal design of experiments for parameter learning in image segmentation.Signal Processing 2013;93:1694-1704.
[32]International Conference on Harmonization.Guideline on validation of analytical procedures:text and methodology Q2(R1)[EB/OL].London:Technical Coordination;2005.
*Corresponding author.Xi’an Jiaotong University Health Science Center,No.76,Yanta East Road,Xi’an 710061,China.Tel./fax:+86 029 82655382.
E-mail address:fuqiang@mail.xjtu.edu.cn(Q.Fu).
Peer review under responsibility of Shenyang Pharmaceutical University
Production and hosting by Elsevier
1818-0876/$-see front matter © 2014 Shenyang Pharmaceutical University.Production and hosting by Elsevier B.V.All rights reserved.
http://dx.doi.org/10.1016/j.ajps.2013.12.007
杂志排行

Asian Journal of Pharmacentical Sciences的其它文章
- Novel chemical permeation enhancers for transdermal drug delivery
- Current prodrug strategies for improving oral absorption of nucleoside analogues
- Application of sialic acid/polysialic acid in the drug delivery systems
- Mesoporous carbon as a carrier for celecoxib:The improved inhibition effect on MDA-MB-231 cells migration and invasion
- Development and evaluation of lafutidine solid dispersion via hot melt extrusion:Investigating drug-polymer miscibility with advanced characterisation
- Rapid and sensitive analysis of cyclobenzaprine by LC-MS/MS:Application to a pharmacokinetic study of cyclobenzaprine in dog