Development and evaluation of lafutidine solid dispersion via hot melt extrusion:Investigating drug-polymer miscibility with advanced characterisation
2014-04-20
Department of Pharmaceutical Sciences and Technology,Institute of Chemical Technology,Nathalal Parekh Marg, Matunga,Mumbai 400019,Maharashtra,India
Development and evaluation of lafutidine solid dispersion via hot melt extrusion:Investigating drug-polymer miscibility with advanced characterisation
Ritesh Fule*,Purnima Amin
Department of Pharmaceutical Sciences and Technology,Institute of Chemical Technology,Nathalal Parekh Marg, Matunga,Mumbai 400019,Maharashtra,India
A R T I C L E I N F O
Article history:
Received 30 October 2013
Received in revised form
25 November 2013
Accepted 16 December 2013
Available online 31 December 2013
Lafutidine
Solid dispersion
Hot melt extrusion
Dissolution rate
Raman spectroscopy
Atomic force microscopy
In current study,immediate release solid dispersion(SD)formulation of antiulcer drug lafutidine(LAFT)was developed using hot melt extrusion(HME)technique.Amphiphilic Soluplus®used as a primary solubilizing agent,with different concentrations of selected surfactants like PEG 400,Lutrol F127(LF127),Lutrol F68(LF68)were used to investigate their inf l uence on formulations processing via HME.Prepared amorphous glassy solid dispersion was found to be thermodynamically and physicochemically stable.On the contrary,traces of crystalline LAFT not observed in the extrudates according to differential scanning calorimetry(DSC),X-ray diffraction(XRD),scanning electron microscopy(SEM)and Raman spectroscopy.Raman micro spectrometry had the lowest detection limit of LAFT crystals compared with XRD and DSC.Atomic Force microscopy(AFM)studies revealed drugpolymer molecular miscibility and surface interaction at micro level.1H-COSY NMR spectroscopy conf i rmed miscibility and interaction between LAFT and Soluplus®,with chemical shift drifting and line broadening.MD simulation studies using computational modelling showed intermolecular interaction between molecules.Dissolution rate and solubility of LAFT was enhanced remarkably in developed SD systems.Optimized ratio of polymer and surfactants played crucial role in dissolution rate enhancement of LAFT SD. The obtained results suggested that developed LAFT has promising potential for oral delivery and might be an eff i cacious approach for enhancing the therapeutic potential of LAFT.
© 2014 Shenyang Pharmaceutical University.Production and hosting by Elsevier B.V.All rights reserved.
1. Introduction
Pharmaceutical drug development research using hot melt extrusion(HME)has attracted increasing attention as novel strategy to produce delivery system with enhanced bioavailability as well as solubility of dissolution rate limited APIs [1-3].This technology employs the application of high shear and high temperature to formulate drug-polymer molecularly dispersed systems,can be termed as solid dispersions(SD)or solid solutions[4].HME is an industrially scalable continuous manufacturing technique without the necessities of additional drying or process fragments[5].The distinctiveness of the procedural features allows the fabrication of various drug delivery systems.HME technology has many advantages over traditional processing techniques such as spray drying or coevaporation,which involves organic solvents[6].Homogeneousmono-phasesystemswiththedrugmolecularly dispersed in the polymer matrix,is challenging delivery,as such systemsare intrinsically metastable[7].The formationof melt extrusion involves the exchange of heat energy during HME process and followed by instant cooling of the melt which affects thermodynamic and kinetic properties of forming solid dispersion variance[8].Use of highly water soluble carrier in solid dispersion always increases the chances of crystallization due to swelling behavior when comes in contact with the aqueous GI f l uid[9].Therefore,surface active agents or surfactants used as inhibitors for recrystallization. HME has the unique property to maintain the amorphous state of the drug after the formation of solid dispersion. Literature cited various methods for preparing amorphous solid dispersion such as melt method,solvent evaporation, cyclodextrin inclusion complex,cryo milling which explained the importance of solid dispersion type of formulation strategy[10].
Lafutidine(LAFT)anewlydevelopedhistamineH2-receptor antagonist,inhibits daytime(i.e.,postprandial)as well as nighttime gastric acid secretion in clinical studies.It is practically insoluble in water and has low bioavailability.LAFT has a very low aqueous solubility,which impairs its dissolution in upper gastric f l uid producing problems to prepared systems [11].Overall,these characteristics hinder its therapeutic application by delaying the absorption rate and thereby onset of action or activity[12].Together solubility,permeability and dissolution rate of a drug are essential factors for determining itsoralbioavailability[13].Literaturereportsgenerallyrevealed the fact that drug materials with a very low aqueous solubility will show dissolution rate limited absorption and hence poor bioavailability.Improvement of aqueous solubility in such a case is a valuable assignment to improve therapeutic eff i cacy [14].However there is no literature on the enhancement of solubility of LAFT by hot melt extrusion method reported. Subsequently there is a need to deliver LAFT in formulation with increased solubility and improved dissolution prof i le.
Forthecurrentstudyweselectedpolyvinylcaprolactam-polyvinyl acetate-polyethylene glycol graft copolymer(Soluplus®)a novel polymer with amphiphilic properties and explored its solubilizing potential using HME technology. Soluplus®has been especially developed for hot melt extrusion process. It offers exceptional capabilities for solubilization of BCS class II and class IV drugs,with the extensive possibility of making SD by hot-melt extrusion[15]. Its bulk density is low and has high molecular weight with excellent f l ow properties.The prime objective was to prepare stable SD systems of low Tgand water insoluble drug LAFT using an optimized ratio of drug-polymer-surfactant blends [16].The next part involves physicochemical characterization usingvariousanalyticaltechniquesto understand the drug-polymer molecular interactions.Six-month stability according to the ICH guideline studies was performed and supported by DSC,XRD,dissolution studies.
2. Materials and methods
2.1. Materials
LAFT was obtainedas a generous gift from Alkem Laboratories Ltd.,India.Soluplus®,a hydrophilic graft copolymer of polyvinyl caprolactam-polyvinyl acetate-polyethylene,Lutrol F127 and Lutrol F68 were kindly donated by BASF Corporation, Mumbai,India(Head off i ce Ludwigshafen,Germany).PEG 400 of analytical grade was procured from Sd.Fine Chemicals, Mumbai,India.All other chemicals used were of analytical grade or equivalent quality.
2.2. Methods
Calculation of solubility parameter(δ),glass transition temperature(Tg)and Flory-Huggins parameter(χ).
As an indicator of the drug-polymer miscibility,values of δ were calculated using the Hoftyzer and vanKrevelen group contribution method described by the following Eq.[17].

where,
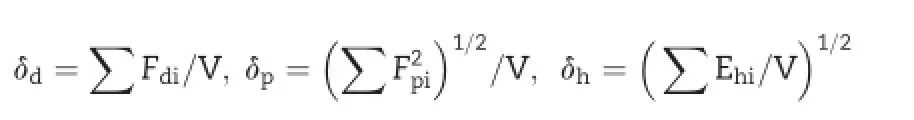
Here i is the groups within the molecule, δ is the total solubility parameter, δdis the contribution from dispersion forces,δpis the contribution from polar interactions,δHis the contribution of hydrogen bonding,Fdiis the molar attraction constant due to molar dispersion forces,Fpiis the molar attraction constant due to molar polarization forces,Ehiis the hydrogen bonding energy and V is the molar volume.The solubility parameters ofpolymer and surfactant combinations were calculated using the following Eq.

where Vf is the volume fraction of each compound.
Miscibility of the drug with the polymer can be assessed based upon the shift in melting endotherm or Tgof the drug or can be predicted theoretically using the Gordon-Taylor equation based on the Tg,densities,and weight fractions of the components.
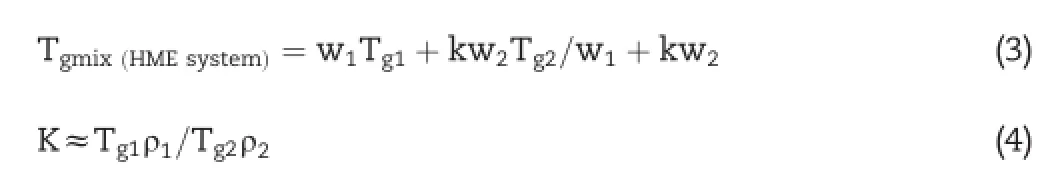
where,Tg1is the glass transition temperature of drug,W1 and W2 are the weight fractions of the components,and K is the parameter calculated from the true densities(ρ1of drug and ρ2of polymer)and Tg2of the amorphous components[18].The true density measurement of the LAFT and Soluplus®were determined in duplicate usinga gas displacement pycnometer (Accupyc 1330;Micromeritics,Norcross,Georgia).
The Flory-Huggins(FH)interaction parameter(χ)was calculated using the following equation:
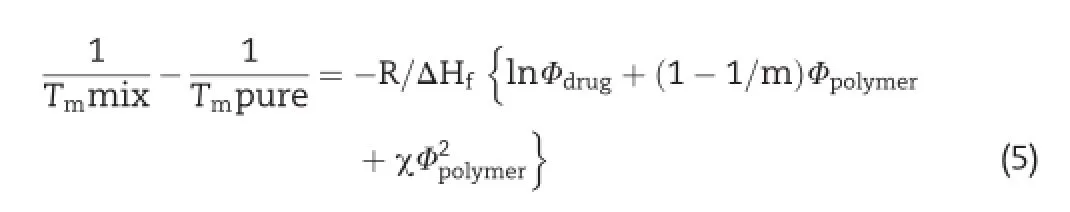
where,Tmmix is the melting temperature of the drug in the presence of the polymer,Tmpure is the melting temperature of the drug in the absence of the polymer, ΔHf is the heat of fusion of the pure drug,m is the ratio of the volume of the polymer to LAFT,and Φ drug and Φ polymer are the volume fractions of the drug and the polymer,respectively[19].
2.3. Preparation of hot melt extruded solid dispersion
Single screw extruder system was used for the hot melt extrusion process manufactured by S.B.Panchal,Mumbai.A die with 2 mm bore diameter was selected based on the prescreening of different dies to obtain uniform extrudes.LAFT and Soluplus®in 1:1 ratio for the batch size of 30 g mixed togetherusing mortarpestlefor 4-5 min.Afterthat,this blend mixture was poured through the hopper on the rotating screw with constant feeding rate;with screw speed of 50 rpm.The extruder temperature was set at 84°C initially(optimized early).The mixture takes about 3 min to form molten mass betweenwallsofthescrewandextruderbarrel.Residencetime was about 15-20 min for LAFT-Soluplus®mixture blends.The similar procedure with different batch size was employed for further drug:polymer combinations(e.g.1:3,1:5,1:7,and 1:9) with different temperature parameters as shown in Table 1. Themeltextrudatesweregrindedandpassedthrougha200 μm sieve.Use of different surfactants such as PEG400,Lutrol F127 andLutrolF68overall14optimizedformulationbatches(LF1to LF14)were practically carried out.In this paper the SD having highest(i.e.50%)drug loading are discussed in terms of physicochemical and dissolution rate characterisation[20].
2.4. Physical state characterization
2.4.1. Differential scanning calorimetry(DSC)and modulated differential scanning calorimetry(M-DSC)
Differential scanning calorimeter(DSC-PYRIS-1,Perkin Elmer, USA)was used to study the drug,polymer and SD crystalline variability.Pure LAFT,Soluplus®,Lutrol®F127,Lutrol®F68 and SD(i.e.4-5 mg)were accurately crimped in aluminium pans and heated at an increment of 10°C/min under a nitrogen purge(20 ml/min)from 0°C to 160°C.During M-DSC accurately weighed samples(4-5 mg)were placed in sealed aluminum pans and a heat-cool-heat cycle applied involving heating from 40 to 260°C at 10°C/min then rapidly cooling to 40°C and then reheating to 260°C at 10°C/min.Both the experiments were performed in a pure dry nitrogen atmosphere using same instrument[21].
2.4.2. Powder X-ray diffractometry
X-ray diffraction pattern were obtained by ADVANCE D8 system with CuKα radiation(Bruker,USA).The recording spectral range was set at 0-40°(2θ)using the Cu-target X-ray tube and Xe-f i lled detector.The voltage 40 kV with current 20 mA was set.The samples were placed in a zero background sample holder and incorporated on a spinner stage.Cu-K′′1 radiation was used as an X-ray source.Soller slits(0.04 rad)were used in the incident and diffracted beam path[22].
2.4.3. FT-IR spectroscopy
Pure LAFT and SD were analysed by using a Fourier transform infrared spectrophotometer model 4100(Spectrum GX-FT-IR, Perkin Elmer,USA).Samples were mixed with dry potassium bromide(dried initially)using a mortar and pestle,compressed to prepare a disk and analyzed over a range4000-400 cm-1.Infrared transform analysis was performed on samples and spectra were generated[23].
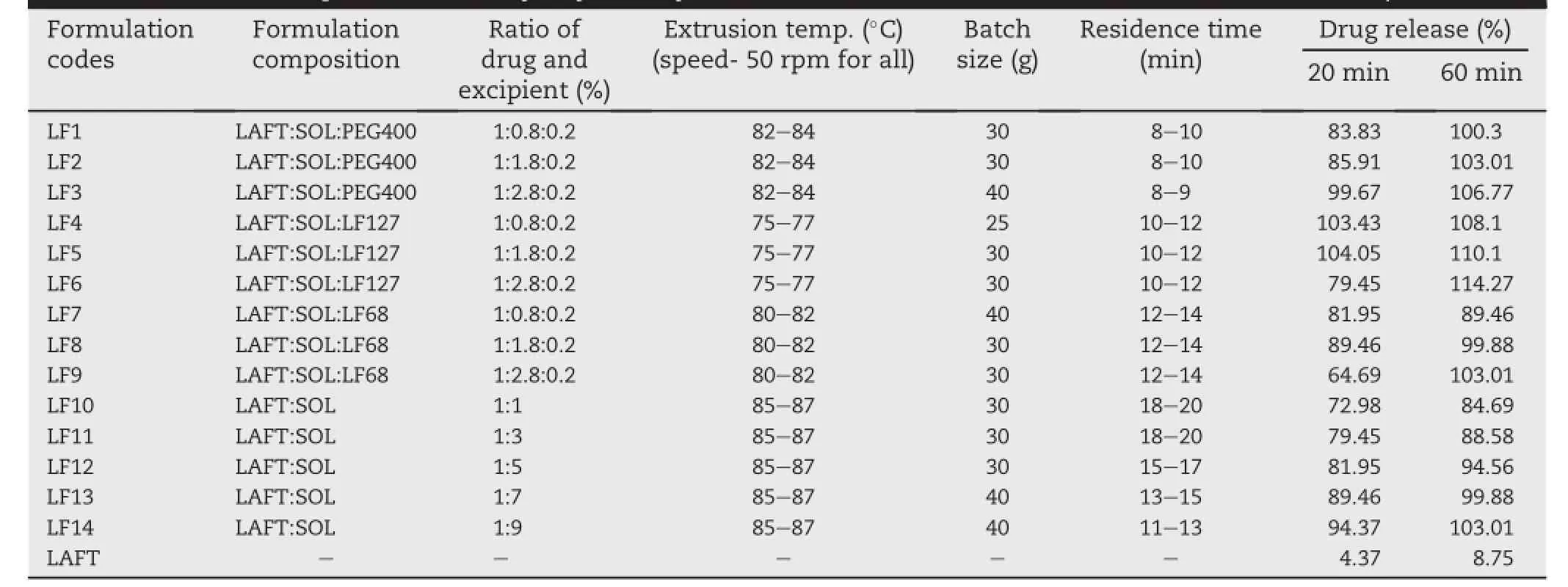
Table 1-Different parameters,ratio of drug to polymer to surfactant and release rate of prepared SD.(Abbreviation: Lafutidine-LAFT,Soluplus®-SOL,Polyethylene Glycol 400-PEG 400,Lutrol F127-LF127,Lutrol F68-LF68).
2.4.4. Scanning electron microscopy(SEM)
The shape and surface morphology of the LAFT powder and LAFT-loaded solid dispersion were examined using XL 30 Model JEOL 6800 scanning electron microscope made in Japan during analysis.Double-sided carbon tape was aff i xed on aluminium stubs over which powder sample of LAFT and prepared SD was sprinkled.The radiation of platinum plasma beam using JFC-1600 auto f i ne coater was targeted on aluminium stubs for its coating to make layer of 2 nm thickness above the sprinkled powder for 25 min.Then,those samples were observed for morphological characterization using a gaseous secondary electron detector(working pressure:0.8 Torr,acceleration voltage:10-30.00 kV).
2.4.5. Raman spectroscopic analyses
The Raman spectra of the SD were recorded with a Lab-RamHR800(Horiba Jovan Yvon)equipped with a 633-nm Ar-Ne laser[24].The laser excitation was focused using a 50 objective(OLYMPAS Corporation)and the scattered light was totally transmitted through the notch f i lter towards the confocal hole and entrance slit of the spectrograph.The stokes-shifted Raman scatter was dispersed using 1800 groove/min grating onto a peltier-cooled changed-coupled device(CCD,Andor Technology PLC)to capture a spectrum. The spectra of SD were recorded more than once and reproducible results were obtained.Raman mapping or imaging of LF4 SD was carried out to understand the drug distribution inside polymer matrix.
2.4.6. Preparation of extrudates for AFM characterisation
JXA-8530F Hyper Probe Electron Probe Micro-analyzer instrument by JEOL was employed for AFM analyses.Freshly fracturedextrudatesonmicroscopic glass slideswere mounted on the micrometre positioning stage of a Dimension Icon AFM with accelerating voltage of 1-30 kV.Probe current range kept between 10 pA and 200 pA and back scattered electron images were obtained.Obtained images were scanned for maximum resolution and magnif i cation to generate best microscopic images[25].
2.4.7. Molecular modelling interaction studies
The monomer unit structures of Soluplus,Lutrol F127,Lutrol F68 and LAFT were constructed by using Gaussian programme in Schrodinger®,maestro software programme,USA.The energy minimization,docking and MD-simulation studies of differentconformationsofdrug-polymerwere run to understand the structural interaction and to identify most stable conformation of drug with polymer[26,27].
2.4.8. 1H-COSY NMR analyses
1H-COSY NMR experiments were carried out on prepared SD (LF4)powder using a Varian Mercury Plus 300 NMR spectrometer operated at 300 MHz with cross polarization contact time of 1 ms,pulse repeat time of 1 s,accumulation of 1000 scans,and high-power 1H-decoupling of 100 kHz during signal acquisition with a-80 to 130°with suitable solvent[28].Suff i cient SD powder sample was dissolved in solvent DMSO and then used for analysis.Sample was spun at a rate of 5 kHz at magic angle with 2D width 4807.7 Hz.5 mm multi nuclear CPMAS probe for solids application was used.Data processing was carried out using sine bell software with FT size 2048 × 2048 and for total time of 65 min.
2.5. HPLC Analyses
LAFT and SD content were determined using a Binary HPLC pump,and 2998 UV Array detector(Agilent Corporation,Milford,Massachusetts)Binary HPLC pump,and 2998 UV Array detector(Agilent Corporation,Milford,Massachusetts)and mixture 0.02 M dihydrogen potassium ortho phosphate and 0.02 M dipotassium hydrogenortho monophosphate(1:1 ratio) with acetonitrile in the ratio of 30:70 adjusted to pH-6 was used as mobile phase.The injection volume was 20 μl and detection was at 215 nm for LAFT and SD[29].
2.6. In vitro dissolution studies
Quantity equivalent to 10 mg of LAFT was weighed and f i lled inside hard gelatine capsules and were used for the dissolution studies further.The LAFT SD and marketed tablets Lafumec®were investigated for their dissolution behavior,in the 900 ml 0.1 N HCl of pH 1.2 as dissolution medium at 37 ± 0.2°C using a USP dissolution apparatus I(Electrolab-DBK,Mumbai,India)at speed of 100 rpm[30].LAFT released from the SD and Lafumec®,characterised by UV absorbance measurement at a wavelength of 286 nm.
2.7. Stability of prepared SD
Prepared SD were kept inside the closed glass vials under controlled temperature environment inside stability chamber (Thermo Lab,India)with relative humidity of(35%,60%,75%) RH and temperature(37°C,40°C,60°C)for stability studies. Samples were removed after 1,3 and 6 months,evaluated for dissolution rate study and compared with those SD testedimmediately after preparation[31].The assay of the drug and SD was evaluated using HPLC at λ =215 nm.
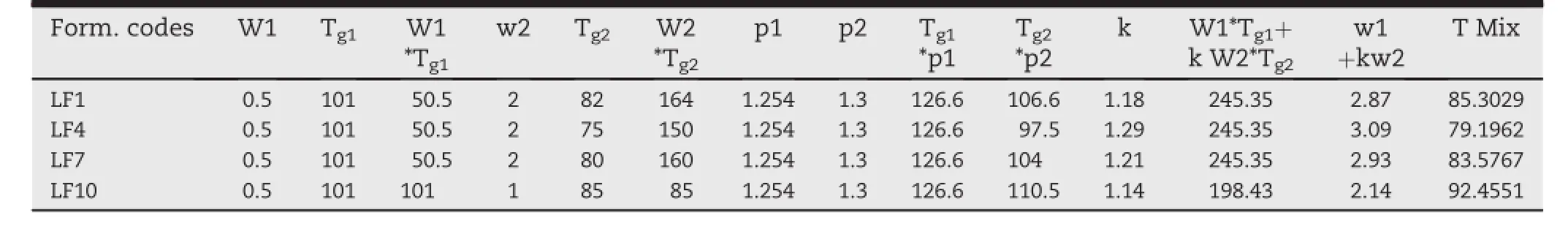
Table 2-Shows the Gordon-Taylor equation calculated Tgmixof SD systems,which are similar(or range of±5°C)to that of experimental HME processing temperature for relevant systems.
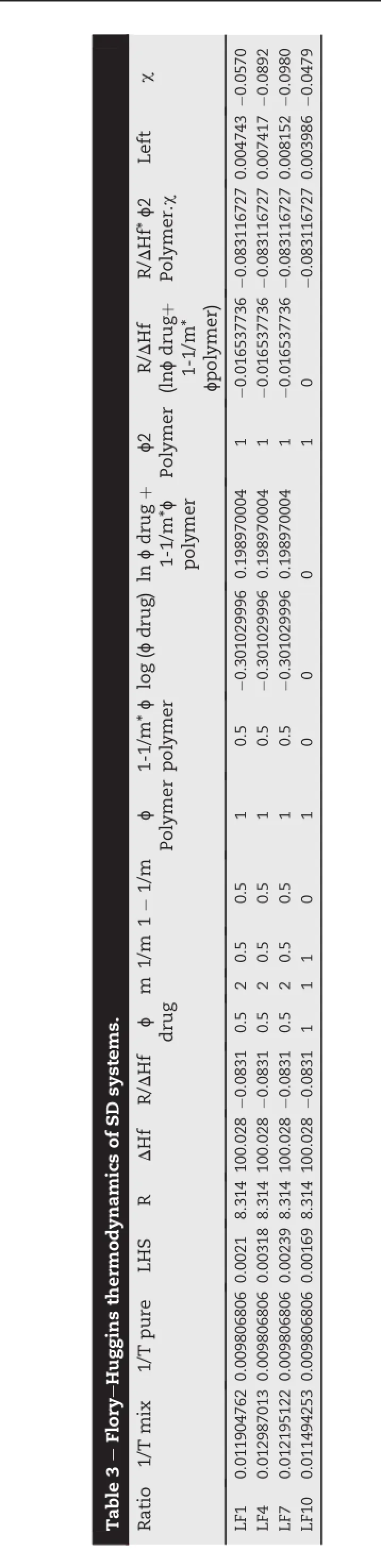
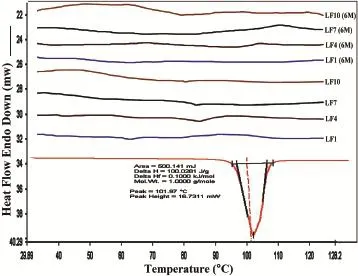
Fig.1-DSC thermograms of LAFT and SD systems.
3. Results and discussion
3.1. Drug-polymer solubility parameter(δ)
It is normally believed that drug-polymer miscibility and phase uniformity at interface depends upon the difference in δ values(Δδ)between two components.If Δδ is less than 7MPa1/2both components are miscible.When Δδ value was 10MPa1/2,incompatibility and phaseseparationbetweendrugpolymer occurs.The solubility parameter for drug and polymer used was calculated using group contribution method (data not shown).The δ of SOL,PEG400,LF127 and LF68 are similar and are close to the reported data.The Δδ values between LAFT-Soluplus and LAFT-Soluplus-surfactant blends were in the range 2.89-5.6 MPa1/2,being less than 7 MPa1/2 indicates likely miscibility.However,the combinations of SOL with PEG400 or LF127 or LF68 appear not to increase the Δδ between LAFT and polymer mixtures,which suggest miscibility enhancement.The Tgvalue of SOL is 72°C.After comparing a series of ratios according to the dispersion state of LAFT and the uniformity of extrudates,drug to polymer ratios were f i nally chosen as the most favourable carriers.The results suggest that Δδ and ΔTgare useful parameters in predicting miscibility and polymer selection[32].
3.2. Gordon-Taylor analysis
Thermal analysis by DSC is the key feature to understand drug-polymer miscibility for the stability of amorphous drug in solid dispersion systems.Incomplete miscibility or reduced solubility can result in the formation of concentrated drug spheres that may lead to recrystallization after production and during stability[33].The Soluplus®showed a Tgof 72°C and of LAFT is 101.97°C.A single Tgwas observed for all the ratios of drug-polymer binary mixtures.According to the Gordon-Taylorequation,ifthedrugandpolymeraremiscible,the binary mixture will exhibit a single Tgthat ranges between the Tgof the pure components and is dependent on the relative proportion of each component shown in Table 2.From the results obtained theoretically by Gordon-Taylor analysis found to be in the similar range of Tg.
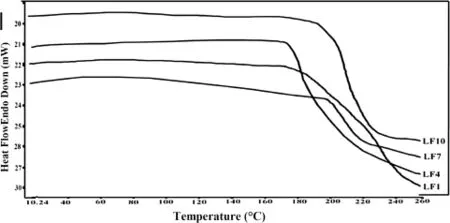
Fig.2-M-DSC thermograms of SD systems.
3.3. Flory-Huggins(FH)modelling
FH modelling suggests that if χ ≥ 0.5/M,then there are slightest amount of unfavourable interactions between the drug,polymer and excipient mixture,which may cause phase separation.From the results,calculated value of FH interaction factor(χ)is not ≥0.5/M which signif i es higher favorable extent of drug-polymer interactions at micro level.This is happened due to reduction of entropy during formation of solid dispersion using HME and also indicates thermodynamic stability of developed SD[34].Adhesive interaction between drug and polymer favoured by the reduction in the Tgof SD systems,which implicates the miscibility of drug and polymer shown in Table 3.
3.4. Solid state characterization
3.4.1. Thermal investigation using DSC and MDSC
Solid-state extruded SD was analyzed using DSC and MDSC. DSC was used to determine the LAFT state in the extruded SD and to identify possible drug-polymer interactions.Fig.1 depicts the thermograms of pure LAFT,which clearly show endothermic sharp peak at 101.97°C respectively.The thermograms of the hot melt extrudates SD showed different thermalbehavior for LAFT.As shown in Fig.1 the drug melting endotherms disappeared completely.The absence of LAFT endotherms suggests either drug solubilization due to the presence of used excipients or being present in an amorphous state.Thus,it was concluded that LAFT converted to its amorphous form during hot melt processing approach used to producethesoliddispersions.MT-DSCstudieswereperformed torecognizethestablenatureofamorphoussoliddispersionof LAFTpreparedbyHME.Comparedtothesharpmeltingpeakof pure LAFT,the endothermic peaks in SD broadened during the f i rst heating cycle and then disappeared in the second heating cycle.This is caused by gradual dissolution of the crystalline drug in the molten polymers and complete conversion to the amorphousstateduringtheDSCheatingprocess.MDSCshows the respective Tgof SD prepared approximately in similar range of temperature which signif i es the amorphous drug nature in SD Fig.2.The physical state of LAFT was further investigated by employing X-ray powder diffraction.The SD prepared after HME convert drug into amorphous stable state.
3.4.2. XRD Analyses
XRD of LAFT consist of sharp multiple peaks,indicating the crystalline nature of the drug with specif i c%crystallinity.In the XRD of LAFT peak intensities observed at(10.23,12.82, 13.12,15.14,17.81,18.12,19.24,21.52,22.34,23.45,24.44,25.62, 27.12,28.22,31.88,41.91,46.43).Characteristic peaks intensities of LAFT observed at 8000,7000,5500,5300 and 4300. In the case of SD(about 2 g)when exposed to X-ray beam, shows disappearance of most of the crystalline endothermic peak and characteristic intensities of LAFT.This indicates complete transformation of crystalline LAFT into amorphous form during HME process.From the XRD studies of both fresh and aged SD systems amorphous nature of LAFT after HME is conf i rmed.The observed few intensity peaks in the diffractograms are attributed to the tablet excipients such as PEG400,Lutrol F127 and Lutrol F68 as shown in Fig.3.The diffractograms indicate that LAFT is in amorphous state(or molecularly dispersed)in the solid dispersions.The XRD results are in good agreement with those of DSC thermograms.
3.4.3. FTIR Analyses
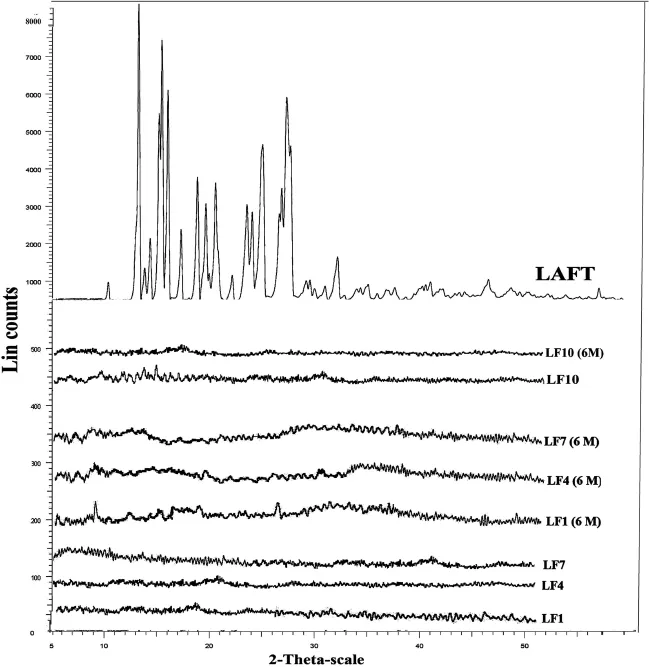
Fig.3-XRD patterns of pure LAFT,LF1,LF4,LF7 and LF10 SD for fresh and aged systems.
Possible interactions between drug and polymer in SD were investigated by FTIR.FTIR spectra of LAFT and SD were examined.FTIR spectrums are properly labelled and shown in (Fig.4A).IR of pure LAFT characteristic sharp peaks of alkene stretching(=C-H and CH2)vibration at 3324.32-3016.48 cm-1and alkane stretching(-CH3,-CH2and-CH)vibration at 2853.73cm-1.AlsoexhibitedC=O stretchat1738.2cm-1dueto saturated ketone and C=O-NH stretching at 1635.90 cm-1.A selective stretching vibration at 1561.57 cm-1and 1525.80 cm-1for primary and secondary amine was also observed.For functional groups like S=O stretch and-C-S stretch showed vibrations at 1041.78 cm-1and 729.57 cm-1respectively.Most of the peaks are observed in the spectral region 785-877 cm-1,570-700 cm-1,and 820-985 cm-1are dueto stretching(bending=C-H and=CH2),-CHdeformation and-CH bending.In the IR spectra of LF1 showed characteristic peaks at 3385.48 cm-1,1737.1 cm-1,1602.19 cm-1, 1384.67 cm-1,1078.46 cm-1,841.65 cm-1and 714.02 cm-1.In theIR spectraofLF4showed characteristicpeaksat 3408.99 cm-1,1736.36 cm-1,1607.10 cm-1,1384.41 cm-1, 1238.92 cm-1,1111.13 cm-1,973.04 cm-1,841.33 cm-1, 715.52 cm-1and 604.15 cm-1.In the IR spectra of LF7 showed characteristic peaks at 3400.80 cm-1, 1731.75 cm-1, 1598.90 cm-1,1384.73 cm-1,1110.72 cm-1,841.16 cm-1, 789.56 cm-1,713.49 cm-1and 608.48 cm-1.In the IR spectra of LF10 showed characteristic peaks at 2913.25 cm-1, 2352.62cm-1,1731cm-1,1614.15cm-1,1555.85cm-1, 1445.07 cm-1,1123.38 cm-1,841.98 cm-1,716.78 cm-1, 604.86 cm-1and 514.30 cm-1.The IR spectra of SD signify the presence of drug and no change in its functional properties. An IR spectrum of Soluplus®represents two characteristic peaks at 2913 cm-1and 1612.39 cm-1.While IR spectrum of unprocessed Lutrol F127 and Lutrol F68 showed characteristic peaks at2870.19 cm-1,1975.36 cm-1,1590.27 cm-1, 1466.88 cm-1,1343.65 cm-1,1112.01 cm-1,962.69 cm-1, 841.98 cm-1,528.39 cm-1and 2884.01 cm-1,1966.16 cm-1, 1592.16 cm-1,1466.26 cm-1,1343.62 cm-1,1121.69 cm-1, 963.15 cm-1,842.63 cm-1,528.95 cm-1respectively.The addition of polymer and surfactant during HME process would not affected LAFT molecule stretching vibrations.Interaction between the polymer and drug in SD mixtures formed molecular dispersions with slight shifting of specif i c intensities compared to pure LAFT IR spectrum.Free hydrogen atoms forms hydrogen bonds with LAFT in the SD possibly.The carbonyl group is more favourable for hydrogen bonding and intermolecular interactions than the nitrogen atom because of steric hindrance.For SD,the-OH stretching bands broadened and the intensity of the bands decreased to minimal, indicating specif i c degree of interaction between the protondonating groups of LAFT and the proton accepting groups in the Soluplus®.IR spectra of used polymer and surfactant are shown in Fig.4B.Also,the IR peak intensities for selective functional groups are represented in Table 4.
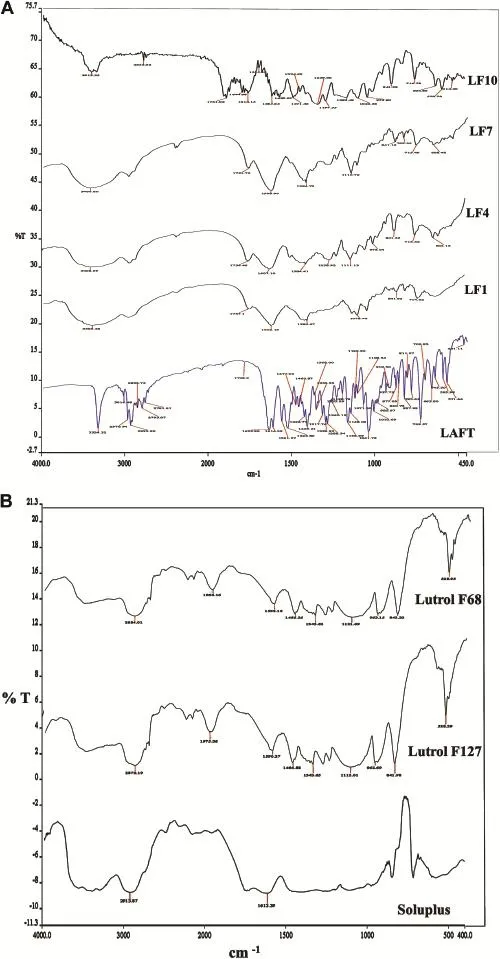
Fig.4-A.Infrared spectroscopic diagrams of pure LAFT and SD systems.B.Infrared spectroscopic diagrams of Polymers.
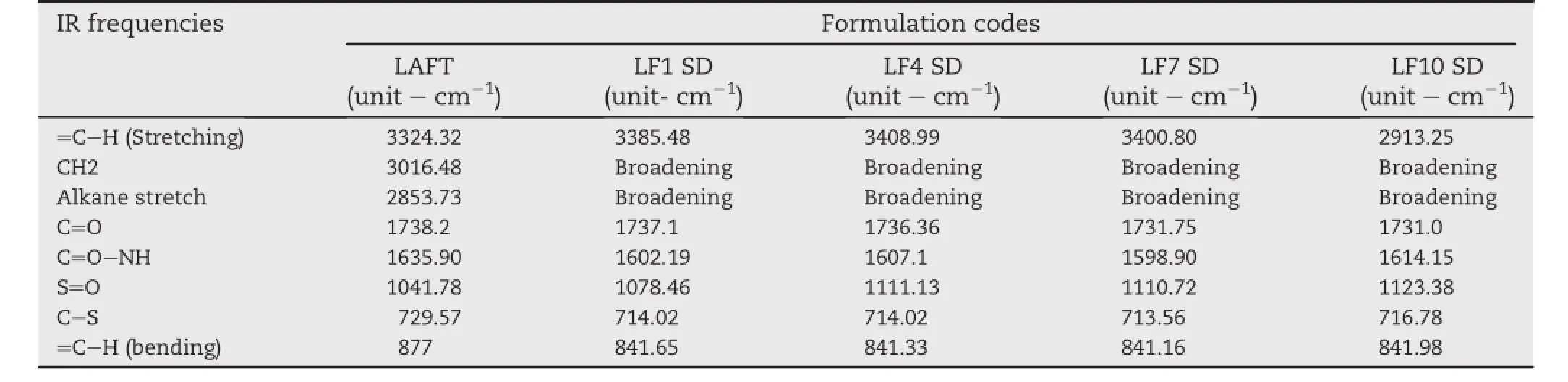
Table 4-Selective IR peak intensities for pure LAFT and SD systems.
3.4.4. SEM Micrographs
Surface micrographs of prepared SD and pure LAFT were determined using SEM technique.The SEM micrograph of pure LAFT it observed large crystalline forms of drug agglomerates with ordered shape and size Fig.5(A).SEM of SD prepared using interaction of drug and polymer chains at micro level.The particle size of combined matrix showed marked decrease in size.The surface characteristics of SD show rough disordered and intact structures,which subsequently help to dissolve drug when comes in contact with aqueous f l uid.However,in SD systems presence of relatively rough surface,suggest that hydrophilic polymer and surfactant were spread uniformly on the surface of the drug also (Fig.5B-E).The LAFT SD appeared to be agglomerated with rough surface owing to the miscibility of drug into polymer.
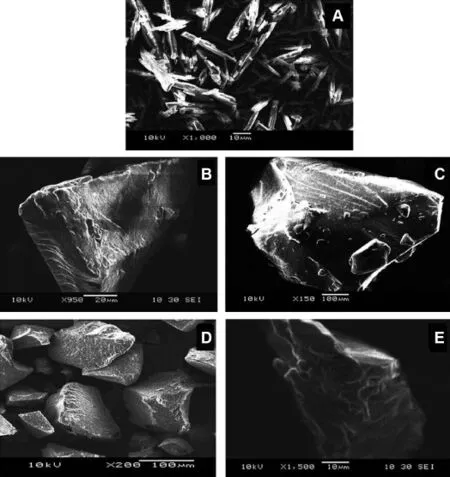
Fig.5-SEM images of Pure LAFT(A),LF1(B),LF4(C),LF7(D),LF10(E).
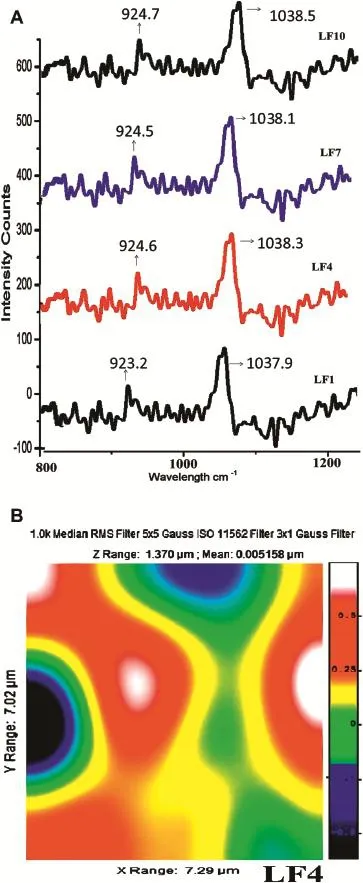
Fig.6-A.Raman spectra of LF1,LF4,LF7,LF10.B.Raman image of LF4.
3.4.5. Raman spectroscopy
With Raman spectroscopy,a laser photon is scattered by sample molecule and loses(or gains)energy during the process.The amount of energy lost is seen as a change in energy (wavelength)of the irradiating photon.This energy loss is characteristic for a particular bond in a molecule.The intensity of spectral features in solution is directly proportional to the concentration of the particular species as shown in Fig.6A.Raman spectra are generally robust to temperature changes.Raman spectroscopy and its mapping technique are useful tools to evaluate crystal and amorphous states, including discrimination of crystalline diastereomer-pairs in soliddispersions as showninFig.6B.Inaddition,by describing the distribution of the drug and the carrier,it could be guessed how drug crystals become amorphous during preparation from the point of view of the distribution of the amorphous form of the drug substance and the carrier.It conf i rms the presence of drug in amorphous form in SD and its uniform distribution[35].Images of the amorphous regions in SDs described by the width at half maximum around 1400 and 1500 cm-1(green area was thought to be amorphous drugs).
3.4.6. AFM Characterisation
Fractured fresh extrudate with smooth surfaces are used for microscopic investigations using AFM.Fracture surfaces were generated at determined fracture points on the outer surfaces of the extrudates.All extrudates had the form of transparent cylindrical rods,2 and 3.5 cm long and of 0.5 cm width were selected,and placed on a sheet of paper.The freshly fractured extrudates weremountedonan opticalglassslide by use ofa 2 component epoxy resin,which hardened within ~5 min. Before the hardening reaction had been completed the extrudate orientation was corrected to get the fracture surface as horizontal as possible.This step is mandatory to enable nondestructive imaging and automated sample changing within Atomic Force Microscope operations[36].Freshly fractured extrudates on microscopic glass slides were mounted on the micrometre positioning stage of a Dimension Icon AFM.Between 10 and 25 regions per sample were programmed to be automatically characterized using the software routine “programmed move”in Tapping Mode.Height,phase,and amplitude images were collected simultaneously,using etched silicon cantileverswith anominalspringconstantof k=40-100 N/m(JEOL AFM Probes).The typical free vibration amplitude was in the range of A=80 nm,the images were recorded with set-point amplitudes corresponding to 60-70% of the free amplitude.Image areas of 10 × 10 mm were recorded at a resolution of 1024 × 1024 pixels.All data were batchprocessed using Scanning Probe Image Processor(SPIP 5.1.1). Height data were plane-corrected by applying a 3rd order polynomial f i t[37].Molecular fracture roughness data as displayed in Fig.7A,consist of image A and B shows cross sectionalsurface roughness calculated from at least 10 images on each sample.The 3D surface image of LF4 showed in image C,which gives morphological surface interactions in detail. The roughness parameters ref l ect the%variation with respect tothetopographymeanheight,whichisshowninimageDand E.It indicates from the AFM analysis that there is high level of surface interaction and amorphousization of drug inside polymer matrix observed in extrudes.Fig.7(B)indicates patterns of AFM images of LF1(F),LF7(G)and LF10(H)extrudes.
3.4.7. Molecular dynamic simulation studies
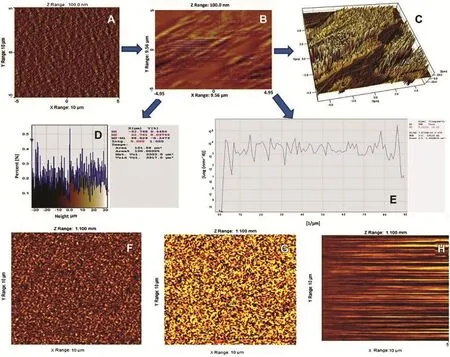
Fig.7-A AFM microscopic image A(LF4),B(LF4 cross sectional),C(LF4 3D surface),D(%area graph)and E(%surface graph). B AFM microscopic images of F(LF1),G(LF7)and H(LF10).
After energy minimization of drug and polymer strong hydrogen bonding interactions were identif i ed.The stable conf i rmation with lowest energy values was optimised and MD simulation dynamics was started.The minimum interaction area from centroid was kept at 4 A°to identify the possible interactions.The energy for stretching,bending, rotational,translational,torsionalkinetic energieswas calculated during this simulation process.The energy of combined system was found to be less than the addition of energies ofindividualmolecules,which signif i es the improved stability of SD formulation.Hydrogen bonding interaction formed with the hydroxyl group of polymers with chlorine group of LAFT.Interactions are formed between the amine group of drug molecule and carbonyl groups of polymers.In both the polymers drug entrapment and interaction was favorable.Both hydroxyl and chlorine group within the LAFT molecule could form strong hydrogen bonds with the monomer of both polymers,which signif i ed by the optimal distance between the H-bond donor and acceptor.MD-simulation studies revealed the possible drug-polymer interactions.The effect of PEG 400,Lutrol F127 and Lutrol F68 on the interaction between LAFT along with Soluplus was found to be favorable.The stable conformations obtained after molecular dynamic simulation showed different geometric arrangement of the molecules.The most stable was fount be Fig.8(B)with lowest energy and highest bonding interaction between drug and polymer.
3.4.8. 1H-COSY NMR investigations
Cross-peaks between all of the protons within a coupling network were observed at protons range of δ 3.3-4.5 ppm with highest number.A small mixing time yields COSY spectra at δ 5.5 ppm.A large mixing time reveals cross-peaks between protons further away in the coupling network at δ 6.5,7,7.5,and 8 ppm are observed.The physical stability of the solid-state drug in amorphous dispersions with amorphous molecular mobility and drug-excipient miscibility was well illustrated by 1H-COSY NMR studies.It is shown in Fig.9 that there is proton-proton coupling observed at various points of resonance which clearly indicates high level of molecular mixing between drug and polymer carrier[38].The entire resonance peaks specif i c for drug and carrier were observed with coupling shifts between due proton resonances.
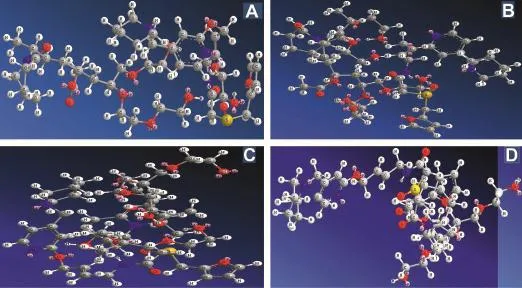
Fig.8-MD simulation conformations of LAFT-Soluplus-PEG 400(A),LAFT-Soluplus-Lutrol F127(B),LAFT-Soluplus-Lutrol F68(C),LAFT-Soluplus(D).
3.5. In vitro dissolution studies
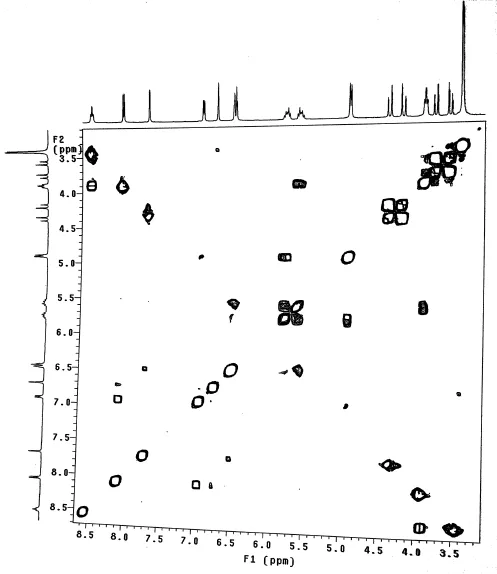
Fig.9-1H COSY NMR spectra of LF4 SD formulation.
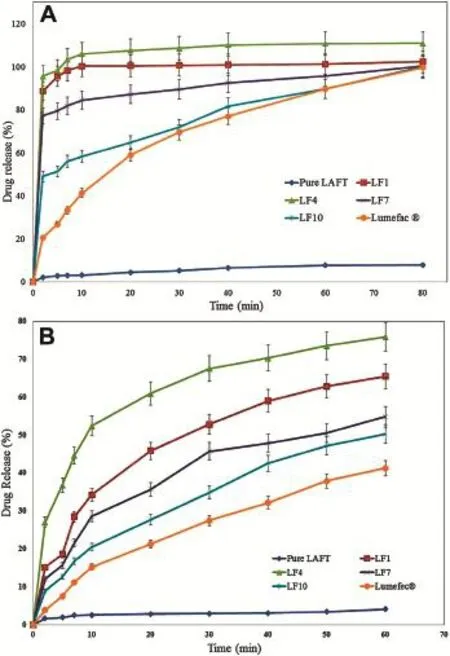
Fig.10-A.In vitro release of pure LAFT,Lumefec®and SD systems in buffer of pH-1.2[mean ± SD(n=3)].B.In vitro release of pure LAFT,Lumefec®and SD systems in distilled water[mean ± SD(n=3)].
Higher apparent drug solubility and improved dissolution prof i les are attributed to the amorphous nature of LAFT in SD system where LAFT is molecularly dispersed in the polymer matrix.SD of LAFT showed disordered morphology.Lattice energy of SD system is due to short-range intermolecular interaction in amorphous system.When drug in SD dissolves then change in lattice energy not destructed by the drug itself so the dissolution rate improved.While,in crystalline form lattice energy has to be destructed for the drug to get dissolve. Hence we don’t observe improved dissolution by simple physical mixing of drug and polymer.Dissolution prof i les of various SD are as shown in Fig.10A and B.The dissolution of the SD with Soluplus®(LF1=100.45%,LF4=110.84%, LF7=95.78%,LF10=84.69%,at the end of T60min)was approximately 11.46,12.58,10.22,9.67 fold higher than pure LAFT respectively.The dissolution of the SD in water (LF1=65.42%,LF4=75.89%,LF7=54.78%,LF10=50.21%at T60min)were approximately 15.87,18.41,13.29,12.18 fold than pure LAFT.The marketed tablet Lafumec®dissolution studies also carried out which is found to be inferior in terms of dissolution as compared to prepared SD.Dissolution of the drug in Soluplus®alone is governed by the carrier,whereas in the case of Soluplus®-surfactant systems,the dissolution rate is governed by solubilization of the polymer to create a hydrotropic environment for the insoluble drug.It was observed that of the Soluplus®-surfactant SD dissolved rapidly.The high dissolution rate of LAFT from the Soluplus®-PEG 400,Soluplus®-Lutrol F127 and Soluplus®-Lutrol F68 dispersion is believed to be due to the drug-polymer molecular intermixing at micro level.The aqueous solubility and dissolution rate ofprepared SD wasalso signif i cantly enhanced.In comparison to pure LAFT,the dissolution rate of physical mixtures was slightly increasedprobably becausethe hydrophilic polymer scan wet the surface of drug particles and acts to solubilize them.The dissolution of extrudates was markedly enhanced with total release occurring within 20 min.This clearly shows that are markable improvement in dissolution performance was achieved by HME[39].From the dissolution prof i les it is evident that HME processing can be employed for the manufacture of LAFT immediate release SD by processing polymer-surfactant combination.The preferred dissolution patterns can be achieved through the drug loading percentageand theextrusionprocess.The extrusionappeared to be an effective approach for the development of diffusion controlled SD of LAFT.
3.5.1. Drug release kinetic evaluation
Drug release prof i les of the optimized LAFT SD are evaluated during dissolution studies.LAFT-SD had shown drug release of 98-104%at the end of 20 min while pure drug(used as reference)released 4-6%at the end of 1 h.The observed transformation in the release pattern strongly indicated the inf l uence of surfactants along with Soluplus®on the dissolution rate enhancement from the SD.The release data was f i tted into various kinetic models like zero order,f i rst order, Higuchi-matrix,Korsemeyer-Peppas,and Hixson-Crowell in order to establish the mechanism of drug release from prepared SD Table 5.SD was found to follow Higuchi release mechanism(r2=0.9973-0.9996)and demonstrated the immediate drug release(n < 0.05)mechanism,which is signif icant with the exposition of more surface area of drug inside SD to the dissolution medium[40].
3.6. Stability on storage
SD is thermodynamically metastable system that favours the conversion of amorphous form in the crystalline form under storage[41].To evaluate the physical state of the drug,the systems were characterized by XRD and DSC after storage for 6 months.The systems were stable during a 6-month period. In the case of SD,no substantial recrystallization was observed by DSC or XRD over the 6 months storage suggesting LAFT is more stable in this formulation.This may be becauseSOL can engage in more extensive hydrogen bonding with LAFT and used surfactants,resulting in less molecular mobility.In addition,there were no signif i cant variations in content of drug and related substances or in dissolution prof i les after storage.Taken together,these results imply that SD formulations are stable over the storage period and that the small quantity of LAFT microcrystals(approximately 2.3%) found after storage has little effect on the stability of the SD. The enhanced physical stability of the SD upon storage is attributed to drug-polymer interactions and solubilization effects of the polymer.Soluplus®-surfactant systems had strong intermolecular interactions,particularly hydrogen bonding between amorphous LAFT and the polymer.These might further reduce the molecular mobility and retarded recrystallization during storage.The stability studies of solid dispersion revealed insignif i cant changes in the stability parameters(p-value <0.05)when kept at 40°C/75%RH,and room temperature respectively at the end of six months.The percent drug entrapments found in the range of 97.4 ± 1.8%-99.2 ± 1.5%in different LAFT SD systems were noted during stability studies.All determinations are performed by using HPLC are mean ± SD(n=3).

Table 5-Order of drug release of SD systems determined by the regression coeff i cients.
4. Conclusion
In the current study it was clearly demonstrated that LAFT immediate release SD formulation can be effectively produced by processing via HME with enhanced solubility and dissolution rate.Novel polymer-surfactant combinations were optimised and stable SD systems were developed successfully.Utilization of Soluplus®along with suitable surfactants offers excellent possibilities to develop stable amorphous solid dispersion.Selective use of surfactants with low concentrations improves process workability,increase melt viscosity with torque reduction,reduce Tgof blend,augments quality of extrudes,reduce residence time of extrusion.The study revealed the importance of suitable carrier and processing technique selection are critical parameters during HME.AFM analyses revealed microscopic surface interaction between drug and polymer.MD simulation studies revealed possible molecular interaction between drug-polymer.1H COSY NMR study conf i rms the molecular mobility and proton-coupling shift at particular resonance.Furthermore,this LAFT-incorporated solid dispersion gave higher dissolution and solubility values compared to the commercial product and pure LAFT powder,indicating that it might improve the oral bioavailability of LAFT in rats.
Acknowledgments
The author is also thankful to UGC(SAP)for providing the research fellowship and Institute of Chemical Technology, ELITE status(Mumbai,India)for providing all facilities and guidance.The authors are thankful to S.A.I.F.department at Indian Institute of Technology,Mumbai for Raman and 1H-COSY NMR experimental help and analyses.
R E F E R E N C E S
[1]Windbergs M,Strachan C,Kleinebudde P.Inf l uence of structural variations on drug release from lipid/polyethylene glycol matrices.Eur J Pharm Sci 2009;37:555-562.
[2]Fule R,Meer T,Amin P,Dhamecha D,Ghadlinge S. Preparation and characterisation of lornoxicam solid dispersion systems using hot melt extrusion technique.J Pharm Invest 2013;43(5)[Article online].
[3]Vithani K,Maniruzzamana M,Slippera I,et al.Sustained release solid lipid matrices processed by hot-melt extrusion. Coll Surf B:Biointerfaces 2013;110:403-410.
[4]Djuris J,Nikolakakis I,Ibricaet S,et al.Preparation of carbamazepine-Soluplus®solid dispersions by hot-melt extrusion,and prediction of drug-polymer miscibility by thermodynamic model f i tting.Eur J Pharm Biopharm 2013;84:228-237.
[5]Fule R,Meer T,Sav A,Amin P.Artemether-Soluplus hot-melt extrudate solid dispersion systems for solubility and dissolution rate enhancement with amorphous state characteristics.J Pharm 2013;1:1-15.
[6]Chokshi RJ,Zia H,Sandhu HK,et al.Improving the dissolution rate of poorly water soluble drug by SD and solid solution:pros and cons.Drug Del 2007;4:33-45.
[7]Tho I,Liepold B,Rosenberg J,et al.Formation of nano/microdispersions with improved dissolution properties upon dispersion of ritonavir melt extrudate in aqueous media.Eur J Pharm Sci 2010;40:25-32.
[8]Stella VJ,Nti-Addae KW.Prodrug strategies to overcome poor water solubility.Adv Drug Del Rev 2007;59:677-694.
[9]Fule R,Meer T,Sav A,Amin P.Solubility and dissolution rate enhancement of lumefantrine using hot melt extrusion technology with physicochemical characterisation.J Pharm Invest 2013;43:305-321.
[10]Sarode A,Sandhu H,Shah N,et al.Hot melt extrusion(HME) for amorphous solid dispersions:predictive tools for processing and impact of drug-polymer interactions on supersaturation.Eur J Pharm Sci 2013;48:371-384.
[11]Itoh H,Naito T,Takeyama M.Lafutidine changes levels of somatostatin,calcitonin gene-related peptide,and secretin in human plasma.Bio Pharm Bull 2002;25:379-382.
[12]Vasconcelos T,Sarmento B,Costa P.SD as strategy to improve oral bioavailability of poorly water soluble drugs. Drug Disc Today 2007;12:1068-1075.
[13]Kalogeras IM.A novel approach for analysing glasstransition temperature vs.composition patterns:application to pharmaceutical compound+polymer systems.Eur J Pharm Sci 2011;42:470-483.
[14]Konno H,Taylor LS.Inf l uence of different polymers on the crystallization tendency of molecularly dispersed amorphous felodipine.J Pharm Sci 2006;95:2692-2705.
[15]Zsombor KN,Attila B,Vajna B,et al.Comparison of electrospun and extruded soluplus®-based solid dosage forms of improved dissolution.J Pharm Sci 2012;101:322-332.
[16]Fule R,Meer T,Sav A,Amin P.Dissolution rate enhancement and physicochemical characterization of artemether and lumefantrine solid dispersions.Int J Drug Del 2012;4:95-106.
[17]Greenhalgh DJ,Williams AC,Timmins PY,et al.Solubility parameters as predictors of miscibility in SD.J Pharm Sci 1999;88:1182-1190.
[18]Dukeck R,Sieger P,Karmwar P.Investigation and correlation of physical stability,dissolution behaviour and interaction parameter of amorphous solid dispersions of telmisartan:a drug development perspective.Eur J Pharm Sci 2013;49:723-731.
[19]Marsac PJ,Shamblin SL,Taylor LS.Theoretical and practical approaches for prediction of drug-polymer miscibility and solubility.Pharm Res 2006;23:2417-2426.
[20]Linn M,Collnot EM,Djuric D,et al.Soluplus®as an effective absorption enhancer of poorly soluble drugs in vitro.Eur J Pharm Sci 2012;45:336-343.
[21]Osama AA,David SJ,Andrews GP.Understanding the performance of melt-extruded poly(ethylene oxide)-bicalutamide solid dispersions:characterisation of microstructural properties using thermal,spectroscopic and drug release methods.J Pharm Sci 2012;101:200-213.
[22]Forster A,Hempenstall J,Tucker I,et al.Selection of excipients for melt extrusion with two poorly water-soluble drugs by solubility parameter calculation and thermal analysis.Int J Pharm 2001;226:147-161.
[23]Andrews GP,Osama AD,Kusmanto F,et al.Physicochemical characterization and drug-release properties of celecoxib hot-melt extruded glass solutions.J Pharm Pharmac 2010;62:1580-1590.
[24]Widjaja E,Kanaujia P,Lau G,et al.Detection of trace crystallinityinanamorphoussystemusingRamanmicroscopy and geometric analysis.Eur J Pharm Sci 2011;42:45-54.
[25]Negi J,Chattopadhyay P,Sharma AK,et al.Development of solid lipid nanoparticles(SLNs)of lopinavir using hot selfnano-emulsif i cation(SNE)technique.Eur J Pharm Sci 2013;48:231-239.
[26]Maniruzzaman M,Morgan DJ,Mendham AP,et al. Drug-polymer intermolecular interactions in hot-melt extruded solid dispersions.Int J Pharm 2013;443:199-208.
[27]Machackova M,Tokarsky J,Capkova P.A simple molecular modelling method for the characterization of polymeric drug carriers.Eur J Pharm Sci 2013;48:316-322.
[28]Zhou P,Xie X,Knight DP,et al.Effects of pH and calcium ions on the conformational transitions in silk f i broin using 2D Raman correlation spectroscopy and 13C solid-state NMR. Biochemistry 2004;43:11302-11311.
[29]Sumithra M,Sundaram P,Srinivasulu K.Analytical method development and validation of LAFT in tablet dosage form by RP-HPLC.Int J Chem Tech Res 2011;3:1403-1407.
[30]Parekh R,Patel P,Patel C,et al.Development and validation of UV spectrophotometric method for estimation of LAFT in bulk and pharmaceutical dosage form.Int J Drug Dev Res 2012;4:325-329.
[31]Breitenbach J.Melt extrusion:from process to drug delivery technology.Eur J Pharm Biopharm 2002;54:107-117.
[32]Weuts I,Kempen D,Decorte A,et al.Phase behaviour analysis of solid dispersions of loperamide and two structurally related compounds with the polymers PVP-K30 and PVP-VA64.Eur J Pharm Sci 2004;22:375-385.
[33]Jung JY,Yoo SD,Lee SH,et al.Enhanced solubility and dissolution rate of itraconazole by an SD technique.Int J Pharm 1999;187:209-218.
[34]Leuner C,Dressman J.Improving drug solubility for oral delivery using SD.Eur J Pharm Biopharm 2000;50:47-60.
[35]Saerens L,Dierickx L,Lenain B,et al.Raman spectroscopy for the in-line polymer-drug quantif i cation and solid state characterization during a pharmaceutical hot-melt extrusion process.Eur J Pharm Biopharm 2011;77:158-163.
[36]Lauer M,Siam M,Tardio J,et al.Rapid assessment of homogeneity and stability of amorphous solid dispersions by atomic force microscopy-from bench to batch.Pharm Res 2013;30:2010-2022.
[37]Almeida A,Saerens L,De Beer T,et al.Upscaling and in-line process monitoring via spectroscopic techniques of ethylene vinyl acetate hot-melt extruded formulations.Int J Pharm 2012;439:223-229.
[38]Natalija Z,Obrezaa A,Bele M,et al.Physical properties and dissolution behaviour of nifedipine/mannitol solid dispersions prepared by hot melt method.Int J Pharm 2005;291:51-58.
[39]Fu J,Zhang L,Tingting G,et al.Stable nimodipine tablets with high bioavailability containing NM-SD prepared by hotmelt extrusion.Powder Tech 2010;204:214-221.
[40]Yang J,Gray K,Doney J,et al.An improved kinetics approach to describe the physical stability of amorphous SD.Int J Pharm 2010;384:24-31.
[41]Sathigari SK,Radhakrishnan VK,Davis VA,et al. Amorphous-state characterization of efavirenz-polymer hot-melt extrusion systems for dissolution enhancement.J Pharm Sci 2012;101:3456-3464.
*Corresponding author.Tel.:+91 22 3361 1111,+91 22 3361 2222;fax:+91 22 3361 1020.
E-mail address:riteshphd.ict@gmail.com(R.Fule).
Peer review under responsibility of Shenyang Pharmaceutical University
Production and hosting by Elsevier
1818-0876/$-see front matter © 2014 Shenyang Pharmaceutical University.Production and hosting by Elsevier B.V.All rights reserved.
http://dx.doi.org/10.1016/j.ajps.2013.12.004
杂志排行

Asian Journal of Pharmacentical Sciences的其它文章
- Novel chemical permeation enhancers for transdermal drug delivery
- Current prodrug strategies for improving oral absorption of nucleoside analogues
- Application of sialic acid/polysialic acid in the drug delivery systems
- Mesoporous carbon as a carrier for celecoxib:The improved inhibition effect on MDA-MB-231 cells migration and invasion
- Determination of azithromycin in raw materials and pharmaceutical formulations by HPLC coupled with an evaporative light scattering detector
- Rapid and sensitive analysis of cyclobenzaprine by LC-MS/MS:Application to a pharmacokinetic study of cyclobenzaprine in dog