Current prodrug strategies for improving oral absorption of nucleoside analogues
2014-04-20,
,
aDepartment of Pharmacy,The Fourth Aff i liated Hospital of China Medical University,No.4, Chongshan Eastern Road,Shenyang 110032,China
bSchool of Pharmacy,China Medical University,No.92,The Northern Second Road,Shenyang 110001,China
cDepartment of Pharmaceutics,Shenyang Pharmaceutical University,No.103,Wenhua Road,Shenyang 110016, China
Current prodrug strategies for improving oral absorption of nucleoside analogues
Youxi Zhanga,b,Yikun Gaoc,Xiaojing Wena,b,Haiying Maa,b,*
aDepartment of Pharmacy,The Fourth Aff i liated Hospital of China Medical University,No.4, Chongshan Eastern Road,Shenyang 110032,China
bSchool of Pharmacy,China Medical University,No.92,The Northern Second Road,Shenyang 110001,China
cDepartment of Pharmaceutics,Shenyang Pharmaceutical University,No.103,Wenhua Road,Shenyang 110016, China
A R T I C L E I N F O
Article history:
Received 31 October 2013
Received in revised form
27 November 2013
Accepted 21 December 2013
Available online 5 January 2014
Nucleoside analogues
Nucleoside analogues are f i rst line chemotherapy in various severe diseases:AIDS(acquired immunodef i ciency disease syndrome),cytomegalovirus infections,cancer,etc. However,many nucleoside analogues exhibit poor oral bioavailability because of their high polarity and low intestinal permeability.In order to get around this drawback,prodrugs have been utilized to improve lipophilicity by chemical modif i cation of the parent drug. Alternatively,prodrugs targeting transporters present in the intestine have been applied to promote the transport of the nucleoside analogues.Valacyclovir and valganciclovir are two classic valine ester prodrugs transported by oligopeptide transporter 1.The ideal prodrug achieves delivery of a parent drug by attaching a non-toxic moiety that is stable during transport,but is readily degraded to the parent drug once at the target.This article presents advances of prodrug approaches for enhancing oral absorption of nucleoside analogues.
© 2014 Shenyang Pharmaceutical University.Production and hosting by Elsevier B.V.All rights reserved.
1. Introduction
Nucleoside analogues are synthetic compounds that are structurally similar to natural nucleosides and,as such,are building blocks of nucleic acids.They act either as inhibitors of cellular and viral DNA andRNA polymerases oraschain terminators by incorporating into a growing DNA or RNA strand.Natural nucleosidesareinvolvedinalmostallcellularprocessesandplaya primary role in structural,energetic,regulatory and metabolic functions.Hence,many nucleoside analogues have cellular cytotoxicity with potency against bacteria,fungi,yeast,virusesor neoplastic tissues,which is attributed to their biochemical mode action[1].Currently,nucleoside analogues are supposed to be drugs that are given in f i rst attention in many serious diseasessuchasacquiredimmunodef i ciencydiseasesyndrome (AIDS),hepatitis,cancer,herpes,smallpox,etc[2].Of the approximately40antiviraldrugsformallyapprovedforuse,half are nucleoside or nucleotide analogues[3].Nucleoside drugs usually must be phosphorylated to the corresponding triphosphates by intracellular or viral kinases[4]in order to exert their pharmacological activity.
Transport of nucleoside analogues across the gastrointestinal tract is often mediated by passive diffusion or active transporters(Na+-independent equilibrative transporters and Na+-dependentconcentrativetransporters)[1].However, their physicochemical properties are unsuitable for passive transcellular intestinal absorption.Meanwhile,nucleoside analogues are not natural substrates and show low aff i nity for nucleoside transporters.Hence,oral absorption of nucleoside analogues is often limited[5].
Variety of nucleoside analogues such as ganciclovir(GCV; marketed Cimevan®and Virgan®)(used for the treatment of cytomegalovirus retinitis)or cidofovir(CDV;marketed as Vistide®)[6,7]are,however,notbioavailableafteroral administration.Others like the anti-HIV nucleosides, although orally bioavailable,suffer from unfavourable pharmacokinetics[8-11]that necessitates the frequent oral administration to maintain therapeutical plasma level in human,also resulting in serious side effects such as bone marrow suppression (anaemia and neutropenia)[12], pancreatitis and peripheral neuropathy[13].
Since the polarity entails nucleosides with low permeability and bioavailability,increasing efforts in the literature are focussing on overcoming these diff i culties with nucleotide prodrugs,an approach which improves the lipophicity and eventually releases the parent nucleotide at a specif i c site.In a nucleotide prodrug,nucleoside analogues are usually covalently bonded to the carrier molecule via phosphoester bond, carboxylic ester bond,carbamate bond or amide bond.The sensitivity of these chemical bonds to enzymatic or chemical hydrolysis has a signif i cant impact on the potency of nucleotide prodrugs.The article highlights the progresses of oral prodrug approaches for nucleoside analogues over the last two decades.Within this review,we have discussed several carboxylic ester prodrugs,monophosphate prodrugs and other prodrugs,with a special focus on rational prodrug design and their performances.It aims to provide some evidences for rational design of oral nucleotide prodrugs.
2. Prodrugs of nucleoside analogues
2.1. Carboxylicacidesters prodrugs
Carboxylicacidesters prodrug approach is widely used to improve oral absorption of nucleosideanalogues,in which the hydroxyl group located at the side chain of nucleoside analogues is esterif i ed with organic acid and vice versa.The carboxylicacidesters-type prodrugs usually possess signif icantenhancementin water-solubility,cellmembrane permeability,enzyme stability and bioavailability,etc.
2.1.1. Acyclovir and its prodrugs
Acyclovir(ACV)belongs to BCS III class drugs and possesses activity against human herpes viruses.However,owing to its limited bioavailability(20%),ACV shows moderate antiviral eff i cacy after oral administration.Hence,it is necessary and feasible to design a prodrug for improving oral absorption of ACV.
Valacyclovir(VACV)is the valine ester prodrug of ACV targeting intestinal oligopeptide transporter 1(PepT1)and has been proved to be safe and effective drug(Fig.1).It has been the most successful prodrug targeting PepT1.PepT1 is a proton-coupled transportingprotein and predominantly distributed in the small intestinal epithelial cells.It has become a striking prodrug-designing target recently,since some poorly absorbed drugs can be modif i ed as peptidomimetic prodrugs targeting intestinal PepT1 to improve oral absorption of the parent drug.3′-hydroxyl group of ACV was esterif i ed withL-valine to prepare VACV.VACV has been reported to increase the oral bioavailability of ACV by 3-to 5-fold in humans.The Cmaxvalues was increased signif i cantly from 2.5 μM to 12.0 μM as well as the AUC0-tvalues from 19.7 μM·h to 49.7 μM·h[14,15].Enhanced oral absorption of ACV has been attributed to the hPepT1-mediated intestinal membrane translocation of prodrug VACV[16,17].Recently,the enzyme responsible for hydrolytic activity toward VACV(hVACVase) has been identif i ed and purif i ed from Caco-2 cells.The high expression of hVACVase in the human intestine,kidney,and liver suggest an important role for hVACVase in the bioactivation of VACV in human tissues[18,19].The peptide transport pathway was supposed to be more eff i cient than the amino acid transport system[20].Therefore,several novel water-soluble dipeptide ester prodrugs of ACV were developed for improving ocular and oral absorption of ACV via targeting the peptide transporters[21].The enzymatic stability and permeability of GVACV(Fig.1)(t1/2=108.1 ± 2.4 min, Papp=2.99 ± 0.59 × 10-6cm/s)was comparable with that of VACV(t1/2=123.7 ± 8.3 min,Papp=3.01 ± 0.21 × 10-6cm/s) [22].Interestingly,the oral bioavailability of ACV for GVACV (AUC0-t=416.1 ± 140.9 μg·min/ml)was approximately 2-fold higher than VACV(AUC0-t=208.4 ± 41.2 μg·min/ml),which may result from its higher enzymatic stability in Caco-2 cell homogenates than VACV[23].Moreover,there was extensive metabolism by hepatic f i rst pass-effect of the dipeptide prodrugs as evidenced by the higher levels of ACV observed in the portal vein than in jugular vein.
After the successful attempt of PepT1-targeted prodrug approach,the dipeptidylpeptidase IV(DPPIV/CD26)prodrug strategy was applied to ACV for improved water-solubility and oral bioavailability.DPPIV/CD26 belongs to a unique class of membrane-associated peptidases[24].It is widely distributed on variety of cell membranes,such as various leucocyte cell subsets and several types of epithelial,endothelial,and f i broblastcells.Furthermore,asolubleformoftheenzymehas been detected in cerebrospinal f l uid and plasma at low amounts[25,26].Depending on the site of attachment of the peptide promoiety,both peptidyl amide and ester prodrugs of ACV were prepared.The tetrapeptide amide prodrug 3(Fig.3) and the tripeptide ester prodrug 4(Fig.1)improved the watersolubility more than 17-fold and 9-fold,respectively,compared to ACV(1.29 mg/ml).In contrast with valine ester prodrug of ACV,both the prodrugs were fully stable in PBS. Meanwhile,they could convert to VACV(for 4)or ACV(for 3) upon exposure to purif i ed DPPIV/CD26 or human or bovine serum[27].This result indicates that the DPPIV/CD26 prodrug approach could be useful for increasing the water-solubility of polar drugs and possibly oral absorption.

Fig.1-Names,chemical structures of the carboxylicacidesters prodrugs described throughout the report.The red parts stand for the parent drugs.
2.1.2. Ganciclovir and valganciclovir
Ganciclovir is an acyclic guanosine analogue,which was f i rst used intravenously to treat CMV infection in AIDS patients.To circumvent the inconvenience and risks associated with frequent intravenous administration,an oral formulation has been further developed,because of its low bioavailability (approximately 5%)[28].
Based on the model exemplif i ed by valacyclovir,valganciclovir,anL-valyl-ester prodrug of ganciclovir(Fig.1),has been synthesized.The bioavailability of orally administered valganciclovir rose up to 61%,almost 10 times higher than the parent drug[29].Consequently,once-daily oral administration of 900 mg of valganciclovir was as effective as once-daily intravenous injection of 5 mg/kg of ganciclovir[30].

Fig.2-Monophosphate prodrugs in this review.The red numbers parts represent the parent drugs.
2.1.3. Didanosine and its prodrugs
Didanosine(5′-O-2′-3′-dideoxydidanosine,DDI)is the second anti-HIV drug approved by the FDA,which is well tolerated with chronic administration with rare and usually reversible toxicity[31].However,DDIexhibitspoor bioavailability (20-40%)[1],which necessitates the continuous infusion to maintain therapeutical plasma level in human.Many investigations had focused on the development of DDI prodrugs for improved oral absorption,but none of them were in routine clinical use[32-34].Yan et al synthesized f i ve peptidomimeticderivativesofDDItargetingPePT1.The5′-O-L-valyl ester prodrug of DDI(5′-O-L-valyl-DDI)(Fig.1)demonstrated the highest permeability in Caco-2 cell model and was selected as the optimal candidate for further studies.The oral absolute bioavailability of DDI was improved from 7.9%to 47.2%after 5′-valyl prodrug orally administered to rats at a dose of 15 mg/kg.It was reported that the prodrug could markedly improve DDI acidic stability in SGF,with the t1/2to be 36 min in SGF,while the parent drug could not be detected in the 2 min.The enhanced acidic stability was conf i rmed again by the coadministration with anti-acid agent in the in vivo oral pharmacokinetics.Antacid combination of DDI increased the oral bioavailability by 115.8%,while 5′-O-L-valyl-DDI(antacid combination)only increased by 30.1%[35].
2.1.4. Penciclovir and famciclovir
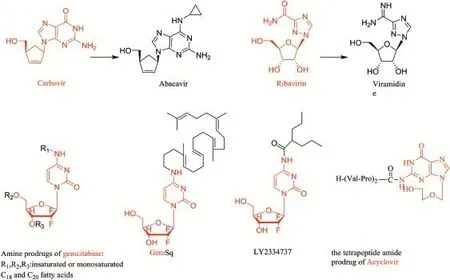
Fig.3-Amide-type prodrugs discussed in this review.The red numbers parts represent the parent drugs.
Penciclovir is an acyclic guanosine nucleoside analogues, which displays a similar spectrum of selectivity and antiviralactivity compared with acyclovir[36].Due to its poor oral bioavailability(F<7%)[37],it is necessary to design an oral alternative of penciclovir.Famciclovir(Fig.1)is a double prodrug containing acetyl diester and 6-deoxy promoieties.It can be eff i ciently bioactived to the parent drug via enzymatic deacetylation and oxidation after oral administration[38,39]. Famciclovir has been proved to be effective for human genital herpes infections and herpes zoster[40].Clinical studies demonstrated the prodrug could be rapidly absorbed and the oral bioavailability of penciclovir rose up to 77%following a single dose of famciclovir[41].In contrast,the acetyl diester of penciclovir did not show any enhancement in oral absorption compared to the parent drug[37].Monocarbonate prodrugs of 6-deoxy penciclovir were also assessed in vivo with the hope of more eff i ciently converting the prodrug to the parent form. Slightly higher or comparable urinary recovery of penciclovir was observed with several monocarbonate prodrugs in mice and rats compared to famciclovir[42].
2.1.5. β-D-2′-Deoxy-2′-f l uoro-2′-C-methylcytidine and RG7128
β-D-2′-Deoxy-2′-f l uoro-2′-C-methylcytidine (PSI-6130), a potent inhibitor of HCV replication in Huh7 replicon assay, exhibits broad activity against HCV without obvious cytotoxicity[43].It entered into clinical trials but showed only modest oral absorption and it was easily degraded to inactive uridine metabolite.In order to get around this drawback,Furman et al prepared a series of bis-isobutyryl ester prodrugs of PSI-6130. It was reported that RG7128(mericitabine;Fig.1)was the most advanced,simple nucleoside prodrug in clinical trials[44]. During Phase I study,RG7128 has shown eff i cacy when applied in patients infected with HCV genotypes 1,2 and 3. RG7128 entered into Phase IIb clinical trials.Early reported data(a 12-week analysis)suggest an average 83%of complete early virological response for the 1000 mg b.i.d./SOC cohort [45].RG7128 is expected to enter the market in the near future.
2.1.6. Cf1743 and its prodrugs
Bicyclic furanopyrimidine nucleoside analogues(BCNAs),a family of highly lipophilic antivirals,display high inhibitory potency and unusual selectivity against varicella zoster virus (VZV)[46].The most potent anti-VZV prototype compound is the p-pentylphenyl BCNA analogue Cf1743,which displays activity against a broad range of VZV isolates at nanomolar concentrations and little or no detectable toxicity at micromolar concentrations[47].However,the very poor aqueous solubility of Cf1743 gives low oral bioavailability(~14%)[48]. Formulation-based strategies were successful in enhancing the water-solubility of Cf1743 but did not have a remarkable impact on oral absorption[48].To solve this problem,Alberto et al synthesized a series of prodrugs of Cf1743(Fig.1)targeting DPPIV/CD26.Alberto et al demonstrated the prodrugs eff i ciently released the parent BCNA drug upon selective conversion by purif i ed DPPIV/CD26 and by soluble DPPIV/ CD26 present in bovine,murine,and human serum.Additionally,the hydrolysis of the prodrugs in the presence of purif i ed DPPIV/CD26,human,murine,and bovine serum was completely blocked,when treating with 2.5 μM vildagliptin(a specif i c inhibitor of DPPIV/CD26).Among the DPPIV/CD26-targeted prodrugs,severalprodrugsshowed signif i cant increase in water-solubility(up to more than 1000-fold) compared to the poorly soluble parent drug.Those prodrugs have been reported to increase the oral bioavailability of Cf1743 by 7-to 5-fold in mice[49].
2.1.7. 3′-Fluoro-2′,3′-dideoxyguanosine and its valyloxypropyl ester prodrug
GlaxoSmithKline Company developed 3′-f l uoro-2′,3′-dideoxyguanosine,which displays activity against HBV.It is phosphorylated by deoxycytidine kinase in vivo to form an active metabolite,3′-f l uoro-2′,3′-dideoxyguanosine triphosphate. This analogue is then incorporated into DNA and prevents DNA reproduction of virus.However,it exhibits low oral bioavailability(only 10% ~ 20%).Hence,its 5′-O-(S)-2-L-valyloxypropyl ester prodrug(Fig.1)was synthesized to improve oral absorption of 3′-f l uoro-2′,3′-dideoxyguanosine.It was reported the oral bioavailability rose up to 50%[50].
2.2. Monophosphate prodrugs
For exerting antiviral activity,the majority of the nucleoside analogues are required to be phosphorylated by a kinase in vivo to form corresponding active metabolites,triphosphates.Since the rate-limiting step in the formation of triphosphate is conversion of nucleoside analogue to its monophosphate,monophosphate ester prodrugs of nucleoside analogues were designed in an attempt to circumvent the initial phosphorylation activation step.Acyclic nucleoside phosphonate(ANP)is a new class of antiviral agents,which does not need initial phosphorylation by viral nucleoside kinases to exert their antiviral effect.Instead,the drugs undergo twophosphorylationreactionsto theiractiveformsby cellular enzymes[51].Consequently,unlike other nucleoside analogues,ANP analogues do not readily lead to virus drug resistance.
However,owing to poor intestinal membrane permeability of the charged molecules,many phosphate prodrugs are not suitable for oral absorption.Hence,prodrug derivatives to mask the ionized phosphate group of nucleosides have been exploited[52,53],but the strategy can be limited because of rapid hydrolysis of the phosphate esters in vivo.However,the problem can be resolved by attaching the phosphonate group to acyclic nucleosidemoietythrougha stableP-C bond,which is resistant to esterase hydrolysis.Prodrugs of the acyclic nucleosidephosphonates(ANP)canmaskthenegative charges on the phosphonate groups and thus improve cellular permeability.
2.2.1. Adefovir and its prodrugs
Adefovir is an acyclic analogues of deoxyadenosine and it displays low oral bioavailability as other ANP analogues due to limited intestinal permeability of the anionic phosphonate moiety[54,55].Hence,various prodrugs of adefovir were designed to mask the charged phosphonate groups and improve oral absorption of adefovir.
It was reported that simple alkyl di-esters or amides prodrugs of adefovirfailed to eff i ciently convert to adefovirin vivo [56].Additionally,monoesters showed poororal bioavailability probably because of the unmasked ionized phosphate groups. In this situation,a bis(pivaloyloxymethyl)ester prodrug ofadefovir(adefovir dipivoxil)(Fig.2)was prepared.The prodrug possesses increased lipophilicity and greaterintestinal permeability than adefovir.Furthermore,it is rapidly convertedtotheparentdruginvivoandleadstoobservablyhigher oral absorption.According to the published data,the oral bioavailabilityofadefovirdipivoxilinhumansroseupto3-and 3.8-fold of free adefovir at dose of 125-500 mg[57,58].Another study with a dose of 10 mg adefovir dipivoxil showed approximately 4-fold increase of oral bioavailability of adefovir[59]. Adefovir dipivoxil is currently licensed for a standard treatment for chronic hepatitis B,particularly in patients with a lamivudine-resistant HBV infection.
2.2.2. Cidofovir and its prodrugs
Cidofovir(HPMPC),an acyclic nucleoside phosphonate,is a potent and selective inhibitor of viral DNA synthesis licensed for the treatment of cytomegalovirus retinitis in AIDS patients [60].Cidofovir itself is a prodrug,which should be transformed to activated triphosphate in vivo.However,due to its polarity of the phosphate group and limited intestinal membrane permeability,HPMPC exhibits low oral bioavailability of <5% and it must be administered by intravenous infusion[7,54]. Meanwhile,cidofovir displays dose-limiting nephrotoxicity due to high concentration in the kidney,which promotes the development of safe cyclic analogue of cidofovir.Cyclic cidofovir(Fig.2)is chemically stable and it can be converted to cidofovir in vivo by a cellular cyclic CMP phosphodiesterase. Compared to intravenous cidofovir,the prodrug has similar antiviral activity but lower potential for nephrotoxicity in humans[61].The oral bioavailability of cyclic cidofovir is also limited,because it can only be absorbed through cell pinocytosisin thegut.Toimproveoralbioavailability ofcidofovirand cyclic cidofovir,ether lipid ester prodrugs were synthesized. These prodrugs were designed to use the lysophosphatidylcholine(LPC)uptake pathway in the small intestine and achieve high oral availability.Esterif i cation of the phosphonatewith severalalkyoxyalkanolsshowed remarkable improvement in oral bioavailability in mice(88-97%)[62-64]. The lipid prodrugs were absorbed intact and converted slowly to the parent drug in tissues.Enhanced antiviral activities of these prodrugs were observed both in vitro and in vivo[65,66]. The improvement in activity was attributed to the increased cellular uptake of cidofovir and intracellular levels of cidofovir diphosphate[67].After that attempt,another prodrug Val-Sercyclic HPMPC was synthesized by Amidon group,which was a promising peptide prodrug targeting puromycin-sensitive aminopeptidase and had been shown to improve the permeability and bioavailability of HPMPC in rodent models.The prodrugwasinitially activated by puromycin-sensitive aminopeptidase to remove theL-valine residue.Subsequent chemical hydrolysis resulted in the generation of cyclic HPMPC[68,69].
2.2.3. Tenofovir and its prodrugs
Tenofovir is structurally similar to adefovir with an extra methyl side chain.As other ANP analogues,poor oral bioavailability of tenofovir(10%)was anticipated in humans. Hence,prodrugs were developed for enhanced oral delivery. Previous clinical experience with adefovir dipivoxil showed thatpivalicaciddischargedfrom theprodrugformed conjugates with carnitine leading to decreased serum carnitine levels[58].Tosolve this problem,a series of prodrugs with alkoxycarbonyloxymethyl groups was designed but with a weak absorption because ofhydrolysis before transmembrane.Meanwhile,carbamate prodrugs also failed to improve bioavailability of tenofovir,probably because of their high enzymatic stability[70].Based on stability,solubility,and enhanced oral absorption(1.5-fold of tenofovir in dogs), bis(isopropyloxycarbonyloxymethyl)ester of tenofovir(tenofovir disoproxil)(Fig.2)was chosen for further study[55,71]. Furthermore,Caco-2 cell transport of the prodrug was increased from 0.1%to 2.7%[55].The oral bioavailability of tenofovir was 25%after oral administration of tenofovir disoproxil fumarate at dose of 300 mg in human and rose up to 39%with food[72].Moreover,tenofovir disoproxil fumarate showed a broad spectrum of antibacterial activities compared to the parent drug and could be applied for the treatment of drug-resistant HIV and HBV infections.
2.2.4. Abacavir and its prodrugs
McGuigan has investigated a series of pronucleotides such as alkyloxyphosphoramidates,phosphorodiamidates,diaryl triesters and aryloxy phosphoramidates.However,the aryloxy phosphoramidates(‘ProTides’)were proved to be the most successful pronucleotides,in which an amino acid ester promoiety was attached to the drug(as an aryl monophosphate or phosphonate)via a P-N bond.The ProTides approach has been applied to many nucleoside analogues, such as 3′-azidothymidine[73],abacavir[74]and tenofovir [75].However,it is diff i cult to obtain optimal antiviral activity of each pronucleotide,the f i ne-tuning of each element(amino acid,ester,and aryl moiety)is required.
Aryloxy phosphoramidates were expected to release the nucleoside monophosphate intracellularly via both chemical and enzymatic mechanisms.The f i rst step is cleavage of the amino acid ester by a carboxyesterase.Subsequently,nucleophilic attack at the phosphorus by the carboxyl group releases the aryloxy group,forming the amino acyl metabolite (AAM).Finally,the amino acid moiety is removed by a phosphoramidase to release the nucleoside monophosphate and an amino acid[76].
The pharmacokinetics and oral bioavailability of abacavir phosphoramidates(Fig.2)were investigated[77].It was found that the pronucleotide was rapidly converted to AAM with a half-life of several minutes,after oral administration of the abacavir methylalaninyl-phosphoramidate.Total exposure to the pronucleotide and its active metabolites was reported to approach that estimated for abacavir at a similar dose, resulting in an overall bioavailability of 50%.
2.3. Amide-type prodrugs
2.3.1. Gemcitabine and its prodrugs
Nucleoside analogues are usually poorly active after oral administration,because of their limited intestinal permeability and rapid metabolism to inactive metabolite in the gut or due to their high f i rst pass metabolism.For example, Gemcitabine(dFdC)is an important anticancer drug that has been licensed for the treatment of non-small cell lung,breast cancer and pancreatic bladder[78].Gemcitabine is extensivelymetabolized to 2′,2′-dif l uoro-2′-deoxyuridine(dFdU)by cytidinedeaminase[79]whichaboundin blood,liver and gut.This has limited dFdC use to the parenteral route in clinical.
Therefore,several oral prodrugs of gemcitabine were designed by coupling acyl chains covalently to the 4-amino group of gemcitabine,in which the 4-amino group was modif i ed with fatty acids[80]or acyclic isoprenoid chain of squalene(GemSq)[81](Fig.3).These lipophilic derivatives of gemcitabine were found to have a slower metabolism in plasma and higher cytotoxicity than gemcitabine.Due to their lipophilicity,these derivatives are also expected to have an increased oral bioavailability compared to gemcitabine.This latest strategy(enhancing lipophilicity)undergoes further development.
LY2334737 is another amide prodrug of gemcitabine(valproic acyl prodrug).Early preclinical studies have shown that LY2334737 is more stable to hydrolysis and leads to enhanced bioavailability by blocking the site of deamination to its uridinemetabolite.This can lead to prolonged systemicexposure ofgemcitabinecomparedto bothIV andoral administration of gemcitabine.Subsequent pharmacokinetic study proved that the prodrug was absorbed mostly intact across the intestinal membrane and then delivered to systemic circulation.The hydrolysis of LY2334737 was relatively slow,leading to sustained release of gemcitabine in vivo[82].Phase I study of oral LY2334737 in Japanese patients with advanced solid tumours demonstrated LY2334737 was tolerated by Japanese patients up to 30 mg/day.The toxicities observed at the 40 mg dose may require the development of alternative dosing schedules [83].
2.3.2. Carbovir and abacavir
Carbovir is a carbocyclic nucleoside analogue with anti-HIV activity.It was abolished as a drug candidate because of poor oral absorption in rats(26%)and monkeys(23%),poor brain penetration,and kidney and cardiac toxicities[84].Abacavir (Fig.3)was a 6-modif i ed carbovir analogues,which is more lipophilic than carbovir with a log P value of 1.22 for abacavir versus-0.62 for carbovir,leading to higher CNS penetration and oral bioavailability(76-100%in animals and 83%in human)[85].After oral administration,abacavir is f i rst phosphorylated to abacavir monophosphate in vivo by adenosine phosphotransferase and then converted to carbovir monophosphate by a cytosolic deaminase,f i nally transformed to carbovir triphosphate[86].The unique activation pathway enabled abacavir to overcome the def i ciencies of carbovir.
2.3.3. Ribavirin and viramidine
Viramidine(Fig.3),a liver-targeting amidine prodrug of ribavirin,is designed to circumvent haemolytic anaemia caused by the parent drug.
Probablydue tothepositivechargeonviramidinemolecule, the uptake of viramidine in red blood cells is less than ribavirin,eventually resulting in a reduction in haematological toxicity[87].The oral bioavailability of viramidine was 66.1%, 43.9%and 61.7%in human,monkey and rat,respectively [88,89].Viramidine is activated to ribavirin in the liver by adenosine deaminase and exhibited a higherliver-toerythrocyte drug ratio,which suggesting better liver-targeting properties.Furthermore,both preclinical and clinical studies showed superiorsafety prof i les ofviramidine compared to the parent drug[90,91].Currently,the prodrug entered in Phase III studies in patients with chronic hepatitis C infection.
3. Conclusion
Nucleotide analogues play an essential role in the treatment of cancer and viruses.Since the rate-limiting step in the formation of triphosphate is conversion of nucleoside analogues to its monophosphate,monophosphate ester prodrugs of nucleoside analogues were designed in an attempt to circumvent the initial phosphorylation activation step.However,both nucleoside analogues and monophosphate ester prodrugs of nucleoside analogues are polar molecules and have limited membrane permeability.Hence,traverse of intestinal epithelial membrane is often limited.Over the past decade,several creative prodrug strategies have been utilized to overcome these limitations.The examples described in this review illustrate the signif i cant research efforts done to improve the oral bioavailability of nucleoside analogues.Traditional prodrug approaches by enhancing lipophilicity have been applied to improve passive diffusion.Prodrugs targeted to PepT1 have been found very useful for enhancing oral absorption of polar drugs.PepT1 has become a promising target since they are highly expressed in the intestine with high capacity and diverse substrate specif i city.Advances in prodrug design have improved the value of nucleoside compounds as anticancer and antiviral agents.The examples described in this article further prove that prodrug approach is an effective strategy for improving oral absorption of nucleoside analogues.
Acknowledgements
This work was f i nancially supported from the Project for Scienceand TechnologyPlan ofLiaoningProvince(No. 2011225020).
R E F E R E N C E S
[1]Balimane PV,Sinko PJ.Involvement of multiple transporters in the oral absorption of nucleoside analogues.Adv Drug Deliv Rev 1999;39:183-209.
[2]Lalanne M,Andrieux K,Couvreur P.Strategies to increase the oral bioavailability of nucleoside analogs.Curr Med Chem 2009;16:1391-1399.
[3]De Clercq E.Antiviral drugs in current clinical use.J Clin Virol 2004;30:115-133.
[4]Rideout JL,Henry DW,Beacham III LM.Nucleosides, nucleotides,and their biological applications.In:Proceedings of the 5th International Round Table,October 20-22,1982. New York:Academic Press;1983.
[5]Li FJ,Maag H,Alfredson T.Prodrugs of nucleoside analogues for improved oral absorption and tissue targeting.J Pharm Sci 2008;97:1109-1134.
[6]Lalezari JP,Friedberg DN,Bissett J,et al.High dose oral ganciclovir treatment for cytomegalovirus retinitis.J Clin Virol 2002;24:67-77.
[7]Cundy KC.Clinical pharmacokinetics of the antiviral nucleotide analogues cidofovir and adefovir.Clin Pharmacokinet 1999;36:127-143.
[8]Collins JM,Klecker RW,Kelley Jr JA,et al.Pyrimidine dideoxyribonucleosides:selectivity of penetration into cerebrospinal f l uid.J Pharmacol Exp Ther 1988;245:466-470.
[9]Gustavson LE,Fukuda EK,Rubio FA,et al.A pilot study of the bioavailability and pharmacokinetics of 2′,3′-dideoxycytidine in patients with AIDS or AIDS-related complex.J Acquir Immune Def i c Syndr 1990;3:28-31.
[10]Klecker RW,Collins Jr JM,Yarchoan R,et al.Plasma and cerebrospinal f l uid pharmacokinetics of 3′-azido-3′-deoxythymidine:a novel pyrimidine analog with potential application for the treatment of patients with AIDS and related diseases.Clin Pharmacol Ther 1987;41:407-412.
[11]Knupp CA,Shyu WC,Dolin R,et al.Pharmacokinetics of didanosine in patients with acquired immunodef i ciency syndrome or acquired immunodef i ciency syndrome-related complex.Clin Pharmacol Ther 1991;49:523-535.
[12]Richman DD,Fischl MA,Grieco MH,et al.The toxicity of azidothymidine(AZT)in the treatment of patients with AIDS and AIDS-related complex.A double-blind,placebocontrolled trial.N Engl J Med 1987;317:192-197.
[13]Yarchoan R,Mitsuya HJ,Pluda M,et al.The National Cancer Institute phase I study of 2′,3′-dideoxyinosine administration in adults with AIDS or AIDS-related complex:analysis of activity and toxicity prof i les.Rev Infect Dis 1990;12:522-533.
[14]Weller S,Blum MR,Doucette M,et al.Pharmacokinetics of the acyclovir prodrug valaciclovir after escalating single-and multiple-dose administration to normal volunte.Clin Pharmacol Ther 1993;54:595-605.
[15]Steingrimsdottir H,Gruber A,Palm C,et al.Bioavailability of aciclovir after oral administration of aciclovir and its prodrug valaciclovir to patients with leukopenia after chemotherapy. Antimicrob Agents Chemother 2000;44:207-209.
[16]De Vrueh RLA,Smith PL,Lee CP.Transport ofL-valineacyclovir via the oligopeptide transporter in the human intestinal cell line,Caco-2.J Pharmacol Exp Ther 1998;286:1166-1170.
[17]Balimane PV,Tamai I,Guo a,et al.Direct evidence for peptide transporter(PepT1)-mediated uptake of a nonpeptide prodrug,valacyclovir.Biochem Biophys Res Commun 1998;250:246-251.
[18]Kim I,Chu X-Y,Kim S,et al.Identif i cation of a human valacyclovirase:biphenyl hydrolase-like protein as valacyclovir hydrolase.J Biol Chem 2003;278:25348-25356.
[19]Puente XS,Lo´pez-Otn C.Cloning and expression analysis of a novel human serine hydrolase with sequence similarity to prokaryotic enzymes involved in the degradation of aromatic compounds.J Biol Chem 1995;270:12926-12932.
[20]Hu M,Subramanian P,Mosberg HI,et al.Use of the peptide carrier system to improve the intestinal absorption of l-αmethyldopa:carrier kinetics,intestinal permeabilities,and in vitro hydrolysis of dipeptidyl derivatives of l-αmethyldopa.Pharm Res 1989;6:66-70.
[21]Nashed Y.Synthesis and characterization of novel dipeptide ester prodrugs of acyclovir.Spectrochim Acta A 2003;59:2033-2039.
[22]Anand BS,Patel J,Mitra AK.Interactions of the dipeptide ester prodrugs of acyclovir with the intestinal oligopeptide transporter:competitive inhibition of glycylsarcosine transport in human intestinal cell.Pharmacology 2003;304:781-791.
[23]Anand BS,Katragadda S,Mitra AK.Pharmacokinetics of novel dipeptide ester prodrugs of acyclovir after oral administration:intestinal absorption and liver metabolism. Pharmacology 2004;311:659-667.
[24]Fox DA,Hussey RE,Fitzgerald KA,et al.Ta1,a novel 105 kDa human T cell activation antigen def i ned by a monoclonal antibody.J Immunol 1984;133:1250-1256.
[25]Mentlein R.Dipeptidyl-peptidase IV(CD26)-role in the inactivation of regulatory peptides.Regul Pept 1999;85:9-24.
[26]Mentlein R.Cell-surface peptidases.Int Rev Cytol 2004;235:165-202.
[27]Diez-Torrubia A,Cabrera S,Castro S,et al.Novel watersoluble prodrugs of acyclovir cleavable by the dipeptidylpeptidase IV(DPP IV/CD26)enzyme.Eur J Med Chem 2013;70:456-468.
[28]Biron KK.Antiviral drugs for cytomegalovirus diseases. Antivir Res 2006;71:154-163.
[29]Leibach FH,Ganapathy V,Ganapathy ME.Transport of valganciclovir,a ganciclovir prodrug,via peptide transporters PEPT1 and PEPT2.J Pharm Sci 2000;89:781-789.
[30]Cvetkovic RS,Wellington K.Valganciclovir:a review of its use in the management of CMV infection and disease in immunocompromised patients.Drugs 2005;65:859-878.
[31]Perry CM,Nobel S.Didanosine-an updated review of its use in HIV infection.Drugs 1999;58:1099-1135.
[32]Singhal D,Ho NF,Anderson BD.Absorption and intestinal metabolism of purine dideoxynucleosides and an adenosine deaminaseactivated prodrug of 20,3-dideoxyinosine in the mesenteric vein cannulated rat ileum.J Pharm Sci 1998;87:569-577.
[33]Anderson BD,Morgan ME,Singhal D.Enhanced oral bioavailability of DDI after administration of 6-Cl-ddP,an adenosine deaminase-activated prodrug,to chronically catheterized rats.Pharm Res 1995;12:1126-1133.
[34]Sriram D,Yogeeswari P,Babu NR,et al.Synthesis and in vitro anti-HIV activities of didanosine prodrugs.J Enzyme Inhib Med Chem 2007;22:51-55.
[35]Yan ZT,Sun J,Chang YN,et al.Bifunctional peptidomimetic prodrugs of didanosine for improved intestinal permeability and enhanced acidic stability:synthesis,transepithelial transport,chemical stability and pharmacokinetics.Mol Pharm 2011;8:319-329.
[36]Boyd MR,Bacon TH,Sutton D,et al.Antiherpesvirus activity of 9-(4-hydroxy-3-hydroxy-methylbut-1-yl)guanine(BRL 39123)in cell culture.Antimicrob Agents Chemother 1987;31:1238-1242.
[37]Morse GD,Shelton MJ,O’Donnell AM.Comparative pharmacokinetics of antiviral nucleoside analogues.Clin Pharmacokinet 1993;24:101-123.
[38]Vere Hodge RA.Famciclovir and penciclovir.The mode of action of famciclovir including its conversion to penciclovir. Antivir Chem Chemother 1993;4:67-84.
[39]Rashidi MR,Smith JA,Clarke SE,et al.In vitro oxidation of famciclovir and 6-eoxypenciclovir by aldehyde oxidase from human,guinea pig,rabbit,and rat liver.Drug Metab Dispos 1997;25:805-813.
[40]Perry CM,Wagstaff AJ.Famciclovir.A review of its pharmacological properties and therapeutic eff i cacy in herpesvirus infection.Drugs 1995;50:396-415.
[41]Pue MA,Benet LZ.Pharmacokinetics of famciclovir in man. Antivir Chem Chemother 1993;4:47-55.
[42]Kim DK,Lee N,Kim YW,et al.Synthesis and evaluation of 2-amino-9-(3-hydroxymethyl-4-alkoxycarbonyloxybut-1-yl) purines as potential prodrugs of penciclovir.J Med Chem 1998;41:3435-3441.
[43]Murakami E,Bao H,Ramesh M,et al.Mechanism of activation of b-d-2′-deoxy-2′-f l uoro-2′-C-methylcytidine and inhibition of hepatitis C virus NS5B RNA polymerase. Antimicrob Agents Chemother 2007;51:503-509.
[44]McHutchison JG,Reddy R,Rodriguez-Torres M,et al.Potent antiviral activity of the nucleoside HCV inhibitor,R7128,inprior IFN nonresponders.In:Presented at:Frontiers in Drug Development in Viral Hepatitis(HEP-DART).Lahaina,Hawaii, USA December 2007.pp.9-13.
[45]Pharmasset.www.pharmasset.com/pipeline/rg7128.aspx.
[46]McGuigan C,Yarnold CJ,Jones G,et al.Potent and selective inhibition of varicella-zoster virus(VZV)by nucleoside analogues with an unusual bicyclic base.J Med Chem 1999;42:4479-4484.
[47]McGuigan C,Barucki H,Blewett S,et al.Highly potent and selective inhibition of varicella-zoster virus by bicyclic furopyrimidine nucleosides bearing an aryl side chain.J Med Chem 2000;43:4993-4997.
[48]McGuigan C,Pathirana RN,Migliore M,et al.Preclinical development of bicyclic nucleoside analogues as potent and selective inhibitors of varicella zoster virus.J Antimicrob Chemother 2007;60:1316-1330.
[49]Alberto D-T,Jan B,Graciela A,et al.Dipeptidyl peptidase IV dependent water-soluble prodrugs of highly lipophilic bicyclic nucleoside analogues.J Med Chem 2011;54:1927-1942.
[50]Leng LY,Cai ZQ,Sun TM.Strategy in the study of nucleoside antiviral prodrugs.Chin J Med Chem 2008;118:310-316.
[51]Bronson JJ,Ho HT,Boeck HD,et al.Biochemical pharmacology of acyclic nucleotide analogues.Ann NY Acad Sci 1990;616:398-407.
[52]Krise JP,Stella VJ.Prodrugs of phosphates, phosphonates,and phosphinates.Adv Drug Deliv Rev 1996;19:287-310.
[53]Meier C.Pro-nucleotides-recent advances in the design of eff i cient tools for the delivery of biologically active nucleoside monophosphates.Synlett 1998;3:233-242.
[54]Wachsman M,Petty BG,Cundy KC,et al.Pharmacokinetics, safety and bioavailability of HPMPC(cidofovir)in human immunodef i ciency virus-infected subjects.Antivir Res 1996;29:153-161.
[55]Naesens L,Bischofberger N,Augustijns P,et al.Antiretroviral eff i cacy and pharmacokinetics of oral bis(isopropyloxycarbonyloxymethyl)-9-(2-phosphonylmethoxypropyl)adenine in mice.Antimicrob Agents Chemother 1998;42:1568-1573.
[56]Starrett JE,Tortolani DR,Russell J,et al.Synthesis, oral bioavailability determination,and in vitro evaluation of prodrugs of the antiviral agent 9-[2-(phosphonomethoxy)ethyl]adenine(PMEA).J Med Chem 1994;37:1857-1864.
[57]Annaert P,Kinget R,Naesens L,et al.Transport,uptake,and metabolism of the bis(pivaloyloxymethyl)-ester prodrugs of 9-(2-phosphonylmethoxyethyl)adenine in an in vitro cell culture system of the intestinal mucosa(Caco-2).Pharm Res 1997;14:492-496.
[58]Barditch-Crovo P,Toole J,Hendrix CW,et al.Antihuman immunodef i ciency virus(HIV)activity,safety,and pharmacokinetics of adefovir dipivoxil(9-[2-(bis-(pivaloyloxymethyl)-phosphonylmethoxyethyl]adenine)in HIV infected patients.J Inf Dis 1997;176:406-413.
[59]Dando TM,Plosker GL.Adefovir dipivoxil.Drugs 2003;63:2215-2234.
[60]Lalezari JP,Stagg RJ,Kuppermann BD,et al.Intravenous cidofovir for peripheral cytomegalovirus retinitis in patients with AIDS:a randomized,controlled trial.Ann Pathol Lab Med 1997;124:362-377.
[61]Mendel DB,Cihlar T,Moon K,et al.Conversion of 1-[((S)-2-hydroxy-2-oxo-1,4,2-dioxaphosphorinan-5-yl)methyl] cytosine to cidofovir by an intracellular cyclic CMP phosphodiesterase.J Antimicrob Agents Chemother 1997;41:641-646.
[62]Ciesla SL,Trahan J,Wan WB,et al.Esterif i cation of cidofovir with alkoxyalkanols increases oral bioavailability and diminishes accumulation in kidney.Antivir Res 2003;59:163-171.
[63]Painter GR,Hostetler KY.Design and development of oral drugs for the prophylaxis and treatment of smallpox infection.TRENDS Biotechnol 2004;22:423-427.
[64]Parker S,Touchette E,Oberle C,et al.Eff i cacy of therapeutic intervention with an oral ether-lipid analogue of cidofovir(CMX001)in a lethal mousepox model.Antivir Res 2008;77:39-49.
[65]Quenelle DC,Collins DJ,Wan WB,et al.Oral treatment of cowpox and vaccinia virus infections in mice with ether lipid esters of cidofovir.Antimicrob Agents Chemother 2004;48:404-412.
[66]Keith KA,Wan WB,Ciesla SL,et al.Inhibitory activity of alkoxyalkyl and alkyl esters of cidofovir and cyclic cidofovir against orthopoxvirus replication in vitro. Antimicrob Agents Chemother 2004;48:1869-1871.
[67]Aldern KA,Ciesla SL,Winegarden KL,et al.Increased antiviral activity of 1-O-hexadecyloxypropyl-[2-14C]cidofovir in MRC-5 human lung f i broblasts is explained by unique cellular uptake and metabolism.Mol Pharmacol 2003;63:678-681.
[68]Eriksson U,Peterson LW,Kashemirov BA,et al.Mol Pharm 2008;5:598-609.
[69]Tehlera U,Nelsona CH,Petersonc LW,et al.Amidona puromycin-sensitive aminopeptidase:an antiviral prodrug activating enzyme.Antivir Res 2010;85:482-489.
[70]Arimilli MN,Kim CU,Dougherty J,et al.Synthesis, in vitro biological evaluation and oral bioavailability of 9-[2-(phosphonomethoxy)-propyl]adenine(PMPA)prodrugs. Antivir Chem Chemother 1997;8:557-564.
[71]Shaw JP,Sueoka CM,Oliyai R,et al.Metabolism and pharmacokinetics of novel oral prodrugs of 9-[(R)-2-(phosphonomethoxy)propyl]adenine(PMPA)in dogs.Pharm Res 1997;14:1824-1829.
[72]Barditch-Crovo P,Deeks SG,Dollier A,et al.Phase I/II trial of the pharmacokinetics,safety,and antiviral activity of tenofovir dioproxil fumarate in human immunodef i ciency virus infected adults.Antimicrob Agents Chemother 2001;45:2733-2739.
[73]McGuigan C,Pathirana RN,Mahmood N,et al.Aryl phosphates derivatives of AZT retain activity against HIV1 in cell lines which are resistant to the action of AZT.Antivir Res 1992;17:311-321.
[74]Balzarini J,Aquaro S,Hassan-Abdallah A,et al. Improved antiviral activity of the aryloxymethoxyalaninyl phosphoramidate(APA)prodrug of abacavir(ABC) is due to the formation of markedly increased carbovir 5′-triphosphate metabolite levels.FEBS Lett 2004;573:38-44.
[75]Lee WA,He GX,Eisenberg E,et al.Selective intracellular activation of a novel prodrug of the human immunodef i ciency virus reverse transcriptase inhibitor tenofovir leads to preferential distribution and accumulation in lymphatic tissue.Antimicrob Agents Chemother 2005;49:1898-1906.
[76]Cahard D,McGuigan C,Balzarini J.Aryloxy phosphoramidate triesters as protides.Mini Rev Med Chem 2004;4:371-381.
[77]McGuigan C,Harris SA,Daluge SM,et al.Application of phosphoramidate pronucleotide technology to abacavir leads to a signif i cant enhancement of antiviral potency.J Med Chem 2005;48:3504-3515.
[78]Toschi L,Finocchiaro G,Bartolini S,et al.Role of gemcitabine in cancer therapy.Future Oncol 2005;1:7-17.
[79]Bergman AM,Pinedo HM,Peters GJ.Determinants of resistance to 2′,2′-dif l uorodeoxycytidine(gemcitabine).Drug Resist Updat 2002;5:19-33.
[80]Myhren F,Borretzen B,Dalen A,et al.EP0986570,2000.
[81]Couvreur P,Stella B,Reddy LH,et al.Squalenoyl nanomedicines as potential therapeutics.Nano Lett 2006;6:2544-2548.
[82]Wickremsinhe E,Bao JQ,Smith R,et al.Preclinical absorption,distribution,metabolism,and excretion of an oral amide prodrug of gemcitabine designed to deliver prolonged systemic exposure.Pharmaceutics 2013;5:261-276.
[83]Yamamoto N,Nokihara H,Yamada Y,et al.Phase I study of oral gemcitabine prodrug(LY2334737)in Japanese patients with advanced solid tumors.Cancer Chemother Pharmacol 2013;71:1645-1655.
[84]Daluge SM,Good SS,Faletto MB,et al.1592U89,a novel carbocyclic nucleoside analog with potent,selective antihuman immunodef i ciency virus activity.Antimicrob Agents Chemother 1997;41:1082-1093.
[85]Chittick GE,Gillotin C,McDowell JA,et al.Abacavir: absolute bioavailability,bioequivalence of three oral formulations,and effect of food.Pharmacotherapy 1999;19:932-942.
[86]Faletto MB,Miller WH,Garvey EP,et al.Unique intracellular activation of the potent anti-human immunodef i ciency virus agent 1592 U89.Antimicrob Agents Chemother 1997;41:1099-1107.
[87]Wu JZ,Lin CC,Hong Z.Ribavirin,viramidine and adenosinedeaminase-catalysed drug activation:Implication for nucleoside prodrug design.J Antimicrob Chemother 2003;52:543-546.
[88]Lin CC,Luu K,Lourenco D,et al.Pharmacokinetics and metabolism of[14C]viramidine in rats and cynomolgus monkeys.Antimicrob Agents Chemother 2003;47:2458-2463.
[89]Lin CC,Xu C,Zhu N,et al.Absorption,metabolism,and excretion of[14C]viramidine in humans.Antimicrob Agents Chemother 2006;50:2368-2373.
[90]Lin CC,Yeh LT,Vitarella D,H,et al.Viramidine,a prodrug of ribavirin,shows better liver-targeting properties and safety prof i les than ribavirin in animals.Antivir Chem Chemother 2003;14:145-152.
[91]Benhamou Y,Pockros P,Rodriguez-Torres M,et al.The safety and eff i cacy of viramidine plus pegylated interferon alpha-2b versus ribavirin plus pegylated interferon alpha-2b in therapy-naı¨ve patients infected with HCV phase 3 results.J Hepatol 2006;44:S273.
*Corresponding author.The Fourth Aff i liated Hospital of China Medical University,No.4,Chongshan Eastern Road,Shenyang 110032, China.Tel./fax:+86 24 62041350.
E-mail addresses:zhangyouxi66@gmail.com(Y.Zhang),cmu4h-mhy@126.com(H.Ma).
Peer review under responsibility of Shenyang Pharmaceutical University
Production and hosting by Elsevier
1818-0876/$-see front matter © 2014 Shenyang Pharmaceutical University.Production and hosting by Elsevier B.V.All rights reserved.
http://dx.doi.org/10.1016/j.ajps.2013.12.006
Oral bioavailability
Prodrug
杂志排行

Asian Journal of Pharmacentical Sciences的其它文章
- Novel chemical permeation enhancers for transdermal drug delivery
- Application of sialic acid/polysialic acid in the drug delivery systems
- Mesoporous carbon as a carrier for celecoxib:The improved inhibition effect on MDA-MB-231 cells migration and invasion
- Development and evaluation of lafutidine solid dispersion via hot melt extrusion:Investigating drug-polymer miscibility with advanced characterisation
- Determination of azithromycin in raw materials and pharmaceutical formulations by HPLC coupled with an evaporative light scattering detector
- Rapid and sensitive analysis of cyclobenzaprine by LC-MS/MS:Application to a pharmacokinetic study of cyclobenzaprine in dog