Shifting balance from neurodegeneration to regeneration of the brain: a novel therapeutic approach to Alzheimer’s disease and related neurodegenerative conditions
2014-04-06NeurodegenerationisoneofthebiggestpublichealthproblemsinmodernsocietyAgeassociatedneurodegenerationwhichisacceleratedseveralfoldinAlzheimerdiseaseADaloneisnotonlyanenormoussocialandeconomicburdentotheaffectedindividual
Neurodegeneration is one of the biggest public health problems in modern society. Age-associated neurodegeneration, which is accelerated several-fold in Alzheimer’s disease (AD) alone, is not only an enormous social and economic burden to the affected individuals and their families, but is also a great scienti fi c challenge. Currently 25-35 million people worldwide suffer from AD, the single largest cause of dementia in middle- to old-aged individuals. These numbers are projected to triple by 2050 if no treatment to prevent or reverse AD is developed.
The two histopathological hallmarks of AD and adults with Down syndrome, who develop AD histopathology in the fourth decade of their lives, are the intraneuronal neuro fi brillary tangles and the extracellular Aβ plaques. The tangles in AD as well as in related neurodegenerative diseases called tauopathies are made up of microtubule associated protein tau modi fied by its abnormal hyperphosphorylation (Grundke-Iqbal et al., 1986a, b). The major components of plaques are 39-43 amino acid fragments, mostly Aβ1-40and Aβ1-42of β-amyloid precursor protein (Glenner and Wong, 1984). Ever since the discoveries of the composition of Aβ plaques and neurofibrillary tangles and implication of those lesions in neurodegeneration, most of the research in the AD fi eld has been focused on inhibition of neurodegeneration by prevention or clearance of these lesions. However, to date, these efforts have not resulted in development of any disease-modifying drug (Iqbal and Grundke-Iqbal, 2010).
Shifting balance from neurodegeneration to regeneration of the brain: a novel therapeutic approach to Alzheimer’s disease and related neurodegenerative conditions
Neurodegeneration is one of the biggest public health problems in modern society. Age-associated neurodegeneration, which is accelerated several-fold in Alzheimer’s disease (AD) alone, is not only an enormous social and economic burden to the affected individuals and their families, but is also a great scienti fi c challenge. Currently 25-35 million people worldwide suffer from AD, the single largest cause of dementia in middle- to old-aged individuals. These numbers are projected to triple by 2050 if no treatment to prevent or reverse AD is developed.
The two histopathological hallmarks of AD and adults with Down syndrome, who develop AD histopathology in the fourth decade of their lives, are the intraneuronal neuro fi brillary tangles and the extracellular Aβ plaques. The tangles in AD as well as in related neurodegenerative diseases called tauopathies are made up of microtubule associated protein tau modi fied by its abnormal hyperphosphorylation (Grundke-Iqbal et al., 1986a, b). The major components of plaques are 39-43 amino acid fragments, mostly Aβ1-40and Aβ1-42of β-amyloid precursor protein (Glenner and Wong, 1984). Ever since the discoveries of the composition of Aβ plaques and neurofibrillary tangles and implication of those lesions in neurodegeneration, most of the research in the AD fi eld has been focused on inhibition of neurodegeneration by prevention or clearance of these lesions. However, to date, these efforts have not resulted in development of any disease-modifying drug (Iqbal and Grundke-Iqbal, 2010).
Loss of neuronal plasticity and unsuccessful neurogenesis
Both in AD and in its animal models the loss of neuronal plasticity is known to precede any overt formation of Aβ plaques and hyperphosphorylated (p) tau neuro fi brillary tangles (Figure 1). Furthermore, the AD brain, in which the hippocampus is the most affected area of the brain and an area of the human brain where neurogenesis is known to occur throughout life, responds to neurodegeneration by stimulating neurogenesis (Figure 2). However, because of the lack of a proper neurotrophic microenvironment of the hippocampus this effort of the AD brain to replace lost neurons with new is unsuccessful (Li et al., 2008). Thus, one potential rational therapeutic approach to AD and other neurodegenerative conditions is to provide a neurotrophic environment in the brain that can materialize into successful neurogenesis and rescue neuronal plasticity de fi cit.
Pharmacologic rescue of neurogenesis and neuronal plasticity de fi cits and cognitive impairment
Taking advantage of the fi ndings that the hippocampal level of the mitogenic factor fibroblast growth factor-2 (FGF-2) is elevated in AD and the ciliary neurotrophic factor (CNTF) can counteract this effect (Chen et al., 2007), we developed a CNTF peptidergic compound, P6 (Chohan et al., 2011). Employing P6 and its more druggable form, P021, a tetrapeptide (DGGL) to which adamantylated glycine was added at the C-terminus, we studied dentate gyrus neurogenesis, neuronal plasticity and cognitive performance in aged Fisher rats and in a transgenic mouse model of both familial AD and familial frontotemporal dementia, the 3xTg-AD mouse model. The aged rat study was reported by Bolognin et al. (2014) and the 3xTg-AD studies by Blanchard et al. (2010) and Kazim et al. (2014).
The Fisher rats are known to become cognitively impaired in old age. We administered P021 by gavage, 500 nmol (289.15 μg)/ kg body weight daily for 3 months to 19-21 month old female Fisher rats. Another group of age-matched female rats was treated identically but with vehicle (saline) only and used as a vehicle-treated control. A group of 2-3 month old female rats treated with vehicle only served as young age controls. During the last week (12thweek) of treatment all three groups of animals were tested for spatial reference memory by Morris Water Maze Task, following which the animals were sacri fi ced and their brains used for immunohistochemical and biochemical studies.
We found that the aged rats were impaired in spatial memory as compared with young adult animals and P021 treatment could rescue this impairment. Immunohistochemical and biochemical analyses showed (i) a marked de fi cit in dentate gyrus neurogenesis in the aged rats which was partly rescued in P021-treated animals; (ii) P021 treatment increased both mRNA and protein expressions of BDNF in the hippocampi of aged rats; (iii) the aged rats displayed a decrease in phosphorylation of CREB which was rescued by P021 treatment; and (iv) P021 treatment increased the levels of synaptic markers, synaptophysin, synapsin-1 and glutamate receptors 2-3, and dendritic marker MAP2 in the aged rats.
Magnetic resonance spectroscopy of the animals before perfusion showed an increase in the production of myoinositol which was rescued in the P021-treated animals. Thus, these studies collectively suggest that pharmacologic enhancement of neurogenesis and neuronal plasticity with a neurotrophic compound is a potential therapeutic approach to cognitive aging and AD.
Bene fi cial effect of pharmacologic neuroregeneration on AD pathology in a transgenic mouse model
In order to determine whether enhancement of regeneration of the brain can be potentially therapeutic both prior to and during the occurrence of AD histopathology, we employed the transgenic 3xTg-AD mouse model. This mouse model expresses human APPSWEand Presenilin-1 M146V knock-in AD and tau P301L frontotemporal dementia mutations, thus a model of both familial AD and familial frontotemporal dementia. These animals show cognitive impairment as early as at ~3 months of age, plaques at about 9-10 months of age and neuro fi brillary tangles at around 12 months (Oddo et al., 2003).
In one study (Blanchard et al., 2010), we administered intraperitoneally P6, 50 nmoles/mouse/day, for six weeks to 6-7 month old female 3xTg-AD mice and genetic background-matched wildtype mice. Mice treated identically to the above except with vehicle (saline) only provided additional controls.
In a test for short term memory, the object recognition task, 3xTg-AD mice displayed impairment to discriminate between a familiar and a new object, accounting for short term memory deficit. Treatment with P6 reversed this impairment. Similarly in a spatial reference memory task employing water maze, 3xTg-AD mice presented delayed performance for learning compared to WT controls, but treatment with P6 reversed this impairment. Treatment with P6 did not induce any side effects as determined by body weight, anxiety, locomotive activity and coordination, and neophobia.
The 3xTg-AD mice revealed a marked deficit in the dentate gyrus neurogenesis which was rescued by P6 treatment. The P6 treatment also rescued de fi cits in markers of neuronal plasticity,i.e., MAP2 and synaptophysin in 3xTg-AD mice.
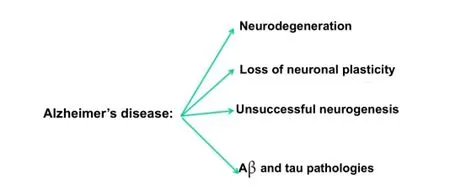
Figure 1 Major features of Alzheimer’s disease pathology.
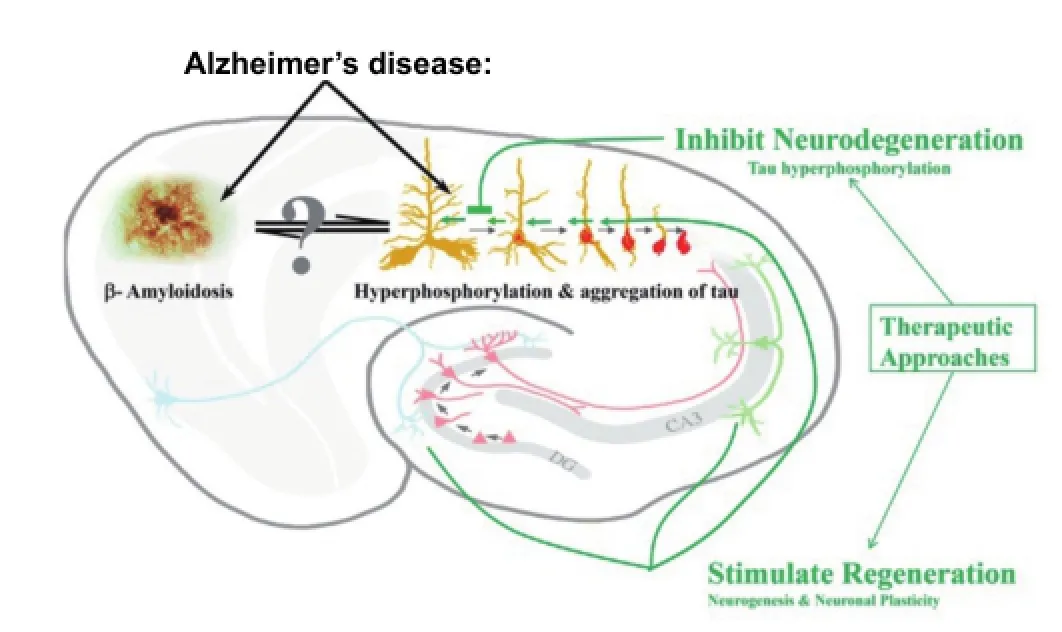
Figure 3 Two major therapeutic approaches to Alzheimer’s disease and related conditions.
In a subsequent study (Kazim et al., 2014. Neurobiol. Dis. In press.) we investigated the effect of 6-12 months treatment with P021 on 9-10-month-old female 3xTg-AD mice. In this study we administered P021, 60 nmol/g diet up to 12 months. Control animals received the same diet but lacking P021. As in the above study with P6, the study with P021 also showed rescue of dentate gyrus neurogenesis and neuronal plasticity de fi cits and of cognitive impairment in 3xTg-AD mice. Moreover, the P021 treatment markedly reduced tau pathology and attenuated the generation but not the clearance of Aβ in 3xTg-AD mice.
Similar to the above described studies, we have also reported rescue of cognitive impairment and neuroplasticity and neurogenesis de fi cits in an experimental rat model of sporadic AD (Bolognin et al., 2012) and in a trisomic Ts65Dn mouse model of Down syndrome (Blanchard et al., 2011). Collectively all these studies have established the proof of principle that shifting balance from neurodegeneration to regeneration of the brain with neurotrophic compounds can rescue Alzheimer type cognitive impairment and several features of the underlying pathology (Figure 3).
Khalid Iqbal, Syed Faraz Kazim, Silvia Bolognin, Julie Blanchard Department of Neurochemistry, Inge Grundke-Iqbal Research Floor, New York State Institute for Basic Research in Developmental Disabilities, Staten Island, NY, USA
Funding:Studies from our laboratory were supported in part by the New York State Office of People with Developmental Disabilities (OPWDD), Zenith Award ZEN-12-241233 from Alzheimer’s Association, and a research grant #20121203 from Alzheimer’s Drug DiscoveryFoundation, New York.
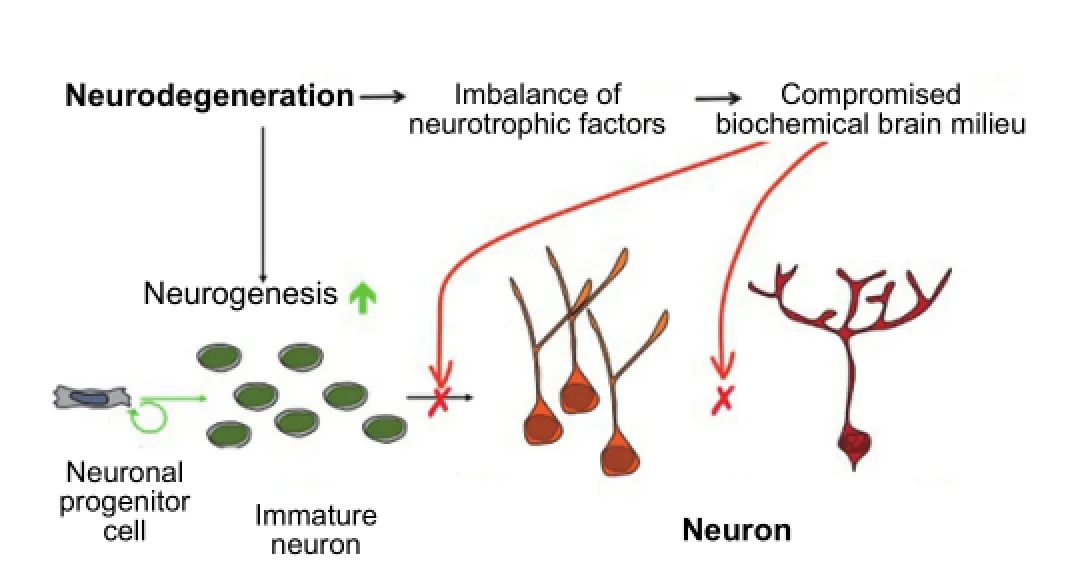
Figure 2 Unsuccessful neurogenesis in Alzheimer’s disease.
Acknowledgements: We would like to thank Janet Murphy who provided the secretarial assistance.
Con flicts of interest: None declared.
Accepted:2014-07-05
Blanchard J, Wanka L, Tung YC, Cardenas-Aguayo Mdel C, LaFerla FM, Iqbal K, Grundke-Iqbal I (2010) Pharmacologic reversal of neurogenic and neuroplastic abnormalities and cognitive impairments without affecting Abeta and tau pathologies in 3xTg-AD mice. Acta Neuropathol 120:605-621.
Blanchard J, Bolognin S, Chohan MO, Rabe A, Iqbal K, Grundke-Iqbal I (2011) Rescue of synaptic failure and alleviation of learning and memory impairments in a trisomic mouse model of Down syndrome. J Neuropathol Exp Neurol 70:1070-1079.
Bolognin S, Blanchard J, Wang X, Basurto-Islas G, Tung YC, Kohlbrenner E, Grundke-Iqbal I, Iqbal K (2012) An experimental rat model of sporadic Alzheimer’s disease and rescue of cognitive impairment with a neurotrophic peptide. Acta Neuropathol 123:133-151.
Bolognin S, Buffelli M, Puolivali J, Iqbal K (2014) Rescue of cognitive-aging by administration of a neurogenic and/or neurotrophic compound. Neurobiol Aging 35:2134-2146.
Chen H, Tung YC, Li B, Iqbal K, Grundke-Iqbal I (2007) Trophic factors counteract elevated FGF-2-induced inhibition of adult neurogenesis. Neurobiol Aging 28:1148-1162.
Chohan MO, Li B, Blanchard J, Tung YC, Heaney AT, Rabe A, Iqbal K, Grundke-Iqbal I (2011) Enhancement of dentate gyrus neurogenesis, dendritic and synaptic plasticity and memory by a neurotrophic peptide. Neurobiol Aging 32:1420-1434.
Glenner GG, Wong CW (1984) Alzheimer’s disease: initial report of the puri fi cation and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun 120:885-890.
Grundke-Iqbal I, Iqbal K, Quinlan M, Tung YC, Zaidi MS, Wisniewski HM (1986a) Microtubule-associated protein tau. A component of Alzheimer paired helical fi laments. J Biol Chem 261:6084-6089.
Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI (1986b) Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A 83:4913-4917.
Iqbal K, Grundke-Iqbal I (2010) Alzheimer’s disease, a multifactorial disorder seeking multitherapies. Alzheimers Dement 6:420-424.
Li B, Yamamori H, Tatebayashi Y, Sha fi t-Zagardo B, Tanimukai H, Chen S, Iqbal K, Grundke-Iqbal I (2008) Failure of neuronal maturation in Alzheimer disease dentate gyrus. J Neuropathol Exp Neurol 67:78-84.
Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM (2003) Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39:409-421.
Khalid Iqbal, Ph.D., Chairman, Department of Neurochemistry, Inge Grundke-Iqbal Research Floor, New York State Institute for Basic Research in Developmental Disabilities, 1050 Forest Hill Road, Staten Island, NY 10314, USA, khalid.iqbal.ibr@gmail.com.
10.4103/1673-5374.139477 http://www.nrronline.org/
Iqbal K, Kazim SF, Bolognin S, Blanchard J. Shifting balance from neurodegeneration to regeneration of the brain: a novel therapeutic approach to Alzheimer disease and related neurodegenerative conditions. Neural Regen Res. 2014;9(16):1518-1519.
杂志排行

中国神经再生研究(英文版)的其它文章
- Neuroprotective effects of berry fruits on neurodegenerative diseases
- Brain structural changes and their correlation with vascular disease in type 2 diabetes mellitus patients: a voxel-based morphometric study
- T cells promote the regeneration of neural precursor cells in the hippocampus of Alzheimer’s disease mice
- Cutaneous sensory nerve as a substitute for auditory nerve in solving deaf-mutes’ hearing problem: an innovation in multi-channel-array skin-hearing technology
- Acrylamide inhibits nerve sprouting induced by botulinum toxin type A
- Transplantation of neurotrophin-3-transfected bone marrow mesenchymal stem cells for the repair of spinal cord injury