遗传性卵磷脂胆固醇酰基转移酶缺乏症肾损害
2014-03-21钟永忠曾彩虹
钟永忠 曾彩虹
病例摘要
现病史40岁女性患者,因“发现尿检异常2月” 于2012-08-27入院。
2012-06-18因月经不调检查发现尿蛋白+++,隐血++,未治疗。7月18日复查尿蛋白2.68 g/24h,隐血++++,白蛋白33.3 g/L,血白细胞计数(WBC) 3.9×109/L,血红蛋白(Hb) 92 g/L,血清肌酐(SCr)正常,服用“百令胶囊”治疗。住外院期间查血液肿瘤标志物、甲状腺功能均正常,自身抗体、补体正常。中段尿培养阴性,消化道超声示脾大,妇科超声示盆腔积液,血压正常,予贝那普利降蛋白尿治疗。本院门诊查尿蛋白定量0.86 g/24h,尿沉渣红细胞计数(RBC) 72万/ml(多形型),肝肾功能正常,血WBC正常,Hb 107g/L,补体正常。病程中患者精神尚可,体力、食欲及睡眠正常,无高血压,无鼻衄、黑便、皮肤淤斑,无发热、皮疹、关节痛,无肉眼血尿史,体重无明显变化,二便正常。既往史无特殊。
家族史患者父亲与母亲为近亲结婚(姑表兄妹)。其父77岁死于肺气肿,母亲健在,患有心脏病,家有4兄妹,大哥体健,二哥及姐姐5岁起视物模糊至今。二哥曾于外院发现角膜成鱼眼状,尿检阴性。其余家庭成员拒绝相关的检查及配合家系调查。
体格检查体温36.6 ℃,脉搏 80次/min,呼吸 18次/min,血压113/62 mmHg,余未发现明显异常。
实验室检查
尿液尿蛋白定量0.63 g/24h,RBC 20万/ml(多形型)。N-乙酰-β-D-氨基葡萄糖苷酶(NAG) 16.78 U/(g·cr),视黄醇结合蛋白(RBP) 0.4 mg/L,溶菌酶<0.5 mg/L,游离轻链33.08 mg/L,游离轻链12.49 mg/L,/λ 2.65。
血常规WBC 3.6×109/L,Hb 91 g/L,血小板计数(PLT) 156×109/L。
血生化胆红素正常,白蛋白40.7 g/L,球蛋白17.4 g/L,谷丙转氨酶(ALT) 12 U/L,谷草转氨酶(AST) 22 U/L,碱性磷酸酶(AKP) 22 U/L,γ-GT 51 U/L,乳酸脱氢酶(LDH) 131 U/L,丁酰胆碱酯酶6 519 U/L,CPK 32 U/L,钙 2.07 mmol/L,葡萄糖4.78 mmol/L,尿素氮(BUN) 5.25 mmol/L,SCr 61 μmol/L,UA 284 μmol/L,钠139.4 mmol/L,钾3.72 mmol/L,氯105.5 mmol/L,磷1.23 mmol/L,TCO225.6 mmol/L,三酰甘油(TG)3.64 mmol/L,总胆固醇(CH)2.22 mmol/L,高密度脂蛋白胆固醇(HDL) 0.16 mmol/L,低密度脂蛋白胆固醇(LDL) 0.35 mmol/L,ApoA1 0.39 g/L,ApoB 0.53 g/L,ApoE 7.02 mg/dl。前白蛋白198 mg/L,C反应蛋白(CRP) 0.1 mg/L,空腹血糖及餐后2h血糖正常。
免疫学ANA、抗dsDNA抗体、ENA多肽抗体谱阴性,补体C3 1.14 g/L,C4 0.22 g/L。IgG 9.560 g/L,IgA 1.370 g/L,IgM 2.700 g/L,IgE 23.4 IU/ml,链球菌溶血素“O”(ASO)<25 IU/ml,类风湿因子(RF)<20。
利用Calbiochem公司的卵磷脂胆固醇酰基转移酶(LCAT)荧光检测试剂盒(EMD Bioscience,San Diego,CA,USA)对患者血浆中的LCAT进行测定发现,患者LCAT活性显著降低。
辅助检查
双肾B超左肾:107 mm×45 mm×53 mm;右肾:103 mm×37 mm×51 mm。
肝、胆、胰、脾B超(1)肝内钙化灶;(2)脾大。
心脏B超(1)二尖瓣钙化;(2)左室舒张功能减低;(3)轻度主动脉瓣反流;(4)轻度二尖瓣反流。
眼科检查裂隙灯检查:双眼角膜边缘变性,白色混浊,中央明,余无异常(图1)。眼底:中心凹反光消失。

图1 眼科裂隙灯检查,患者角膜边缘混浊,似“鱼眼状”
心电图:窦性心律,室性早搏。
胸片:无明显异常。
肾活检病理
光镜29个肾小球中5个球性废弃,余正切肾小球体积增大,系膜区轻度增宽,节段加重,系膜基质增多,系膜区和外周袢肾小球基膜(GBM)见空泡状改变(图2A),偶见系膜溶解,毛细血管袢开放好,节段内皮细胞成对,囊壁增厚、节段分层。PASM-Masson:GBM广泛空泡化呈虫蚀状改变(图2B),部分外周袢分层。肾小管间质慢性病变轻度,小灶性肾小管萎缩、肾小管基膜(TBM)增厚,周围间质小灶性纤维化。小叶间动脉内膜明显增厚,小动脉节段透明变性。肾组织刚果红染色阴性。
免疫荧光肾小球17个。冰冻切片荧光染色IgM+,呈颗粒状弥漫沉积于肾小球系膜区。IgG、IgA、C3、C1q阴性。
电镜电镜下观察2个肾小球。肾小球毛细血管袢开放差,节段内皮细胞剥脱、内皮下区域增宽、疏松。GBM及系膜区较多空泡化改变,少数空泡残留嗜锇性物质,呈层状结构(图3)。GBM厚薄明显不一,薄处约478 nm,厚处1 306~2 276 nm,最厚处达3 540 nm。肾小球系膜区增宽,基膜样物质增多,节段系膜区亦见“嗜锇小体”。肾小管上皮细胞肿胀,胞质内见大量肿胀、变形的线粒体。
最后诊断LCAT缺乏症肾损害。
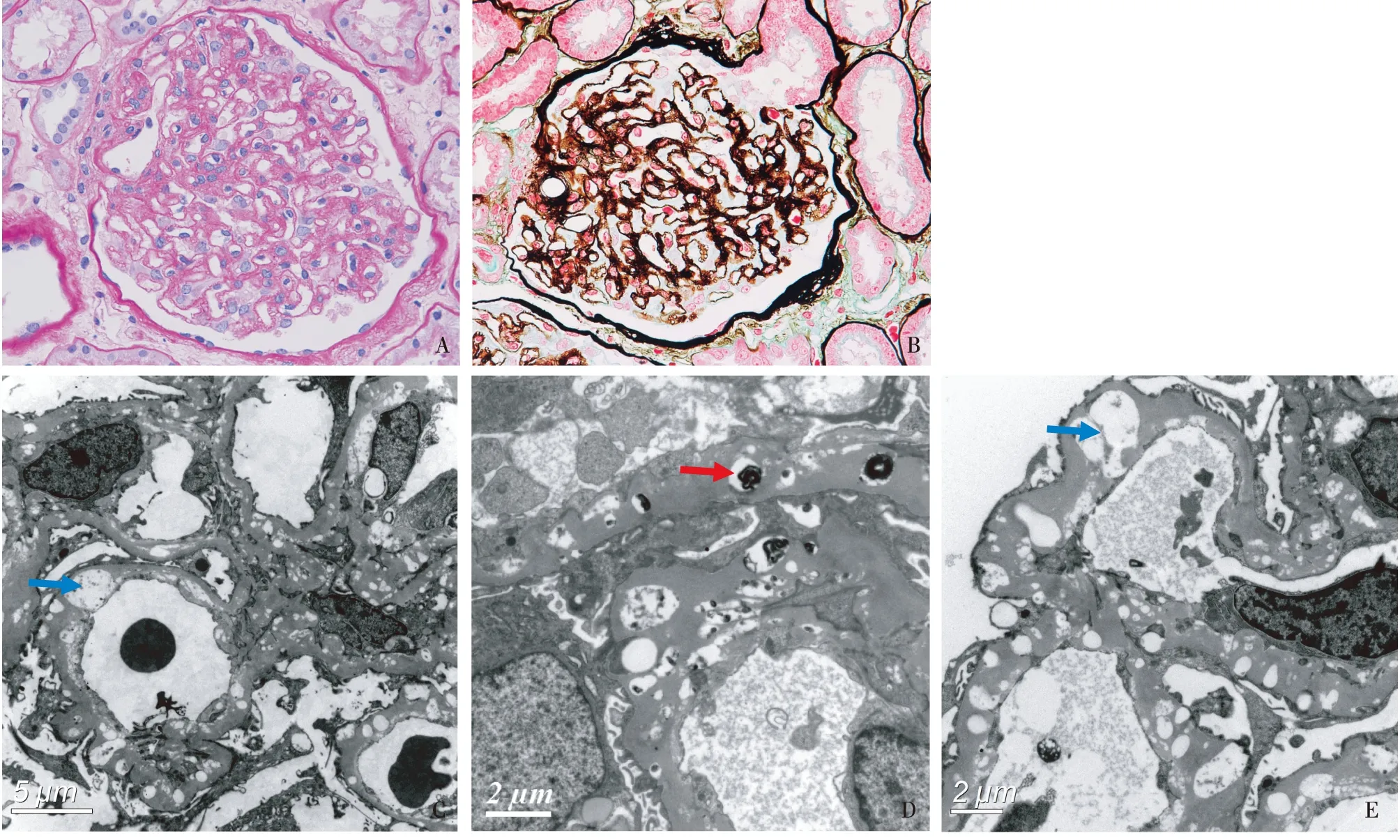
图2 A:肾小球系膜区轻度增宽,系膜区外周袢见空泡状改变(PAS,×400);B:肾小球基膜广泛空泡化伴节段上皮侧钉突样物质形成(PASM-Masson,×400);C~E:肾小球系膜区和肾小球基膜内见大量空泡(↑),大小不均一,部分空泡内残留嗜锇性物质,呈层状改变(↑)(EM)
讨 论
患者肾脏损害以少量蛋白尿和镜下血尿,无高血压,且肾功能正常;光镜下GBM呈空泡或泡沫样改变,似Ⅳ期膜性肾病(MN),但免疫荧光仅见IgM少量沉积于系膜区,电镜下亦未见上皮侧电子致密物及钉突形成,因此不支持MN的诊断。该患者最突出的特点是电镜下观察到GBM内和系膜区广泛分布大小不一的空泡,其中部分残留层状嗜锇性物质,提示这些物质为脂质成分,考虑肾脏损害可能与脂质代谢相关。分析患者脂质代谢异常,主要表现HDL显著降低,血CH下降,TG升高,轻度正色素性贫血。结合患者的父母属近亲结婚(表兄妹),且患者及其二哥存在典型的鱼眼状角膜混浊,考虑遗传性脂质代谢异常相关性疾病,而影响血HDL 成熟及其浓度的主要因素是LCAT,因此高度考虑患者存在LCAT缺乏。对患者血浆LCAT活性检测也证实血浆LCAT几乎无活性。故最终明确诊断为遗传性LCAT缺乏症肾损害。
在认识LCAT缺乏症肾损害临床病理表现之前,应先了解LCAT及LCAT缺乏症。
LCAT的生物学特性及意义LCAT主要是由肝脏合成和分泌,编码基因位于16q21-22号染色体,由6个外显子和5个内含子共4 200个碱基构成。血浆中的LCAT主要存在两种活性:(1)α-LCAT活性,针对HDL中胆固醇底物,需要ApoA1和ApoA4作为其辅助因子;(2)β-LCAT活性是专门针对LDL、VLDL中的胆固醇,不需要辅助因子[1]。LCAT将卵磷脂2位脂酰基转移到HDL3的胆固醇3位羟基上生成胆固醇酯和溶血性卵磷脂,促使盘状的新生HDL3转化为成熟的球状HDL2。LCAT参与胆固醇逆转运,促进组织、细胞内胆固醇的清除,维持细胞内胆固醇的稳态。
遗传性LCAT缺乏症遗传性LCAT缺乏症是一种罕见的脂类代谢异常的常染色体隐性遗传病,根据临床表现形式和LCAT活性分为两种形式:家族性LCAT缺乏症(FLD)和鱼眼病(FED)[1]。FLD是α-LCAT和β-LCAT活性均缺乏或降低,造成血浆中未酯化胆固醇升高,临床表现为角膜混浊、正色素性贫血及肾脏损伤;而FED主要是由于α-LCAT活性的缺乏,β-LCAT活性不变,血浆中未酯化胆固醇正常或轻微上升,临床表现为角膜混浊而肾功能正常,但也有临床表现介于FLD与FED之间的患者。
遗传性LCAT缺乏症主要由于LCAT基因突变所致。目前人类基因突变数据库(HGMD)共收录了97个LCAT基因的突变位点。除了编码区的基因突变造成LCAT酶活性的丧失外,转录或者转录后的异常也能引起LCAT酶活性缺乏[2]。如内含子的突变造成套索形成受阻,影响LCAT mRNA前体的正确拼接,最终影响LCAT的正常合成[3]。对家族性LCAT缺乏症患者的家系筛查中发现,即使相同的基因突变,不同的患者也可存在不同的临床表现[4-6]。这表明,存在其他因素影响着患者的基因型与临床表型之间的关系。该患者未能接受基因检测,尚无法明确其基因突变位点。
此外,也有获得性LCAT缺乏症的报道。(1)产生针对LCAT的自身抗体。Takahashi等[7]报道了1例肾病综合征(NS)患者,LCAT活性严重缺乏。肾脏病理改变除了与遗传性LCAT缺乏症患者类似之外,还表现为MN,免疫荧光和免疫组化染色可发现抗LCAT抗体沿着肾小球毛细血管袢沉积,激素治疗有效,血脂异常明显改善,重复肾活检提示病理损伤较前减轻。Simonelli等[8]也描述了一位淋巴瘤患者体内HDL严重缺乏,血浆LCAT活性极低,产生针对LCAT的特异性抑制性抗体,淋巴瘤治疗好转后,患者的脂质代谢也得到改善。(2) LCAT丢失过多。 Vaziri等[9]认为NS患者滤过膜屏障的破坏,会引起LCAT丢失,造成HDL2/HDL3比例下降。张小瑛等[10]发现NS患者LCAT活性较正常对照组减低。此外LCAT活性降低还可见于慢性肾衰竭[11]、甲状腺功能紊乱[12]等疾病。但获得性LCAT缺乏症患者一般无角膜混浊,且去除诱因之后,LCAT酶活性得到改善。
LCAT缺乏症肾损害的病理改变LCAT缺乏症引起肾损害并不常见。肾小球是主要的受累部位,光镜下可见系膜区轻度增宽,系膜细胞无明显增殖或轻度增多,以系膜基质增多为主,GBM增厚。GBM及系膜区可见泡沫样分布的脂质样物质,呈空泡样或蜂窝状外观,GBM的空泡化病变类似于晚期MN。系膜区可见泡沫细胞,毛细血管袢内相对少见。随着疾病的进展,晚期肾小球可出现节段硬化或者全球硬化。泡沫细胞也可见于肾间质内,早期肾小管病变不明显,晚期可出现灶性肾小管萎缩、TBM增厚、间质纤维化。免疫荧光证实免疫球蛋白和补体阴性,偶尔伴非特异的沉积,肾小球ApoB和ApoE染色可见阳性。电镜是该病诊断的关键,早期脂质沉积主要在GBM上皮侧、基膜内和内皮下区域,随着疾病进展沉积于系膜区[13]。沉积的脂质呈大小不一的空泡状,部分空泡内含层状的匍匐性嗜锇性物质,呈层状或颗粒状,似 “玫瑰花瓣”。动脉内皮细胞和平滑肌细胞胞质内也可见脂质。
LCAT缺乏症肾损害的临床表现遗传性LCAT缺乏症患者早期可能无明显的临床表现,仅血生化发现HDL水平降低,游离胆固醇升高[14]。随着病程进展出现角膜混浊、贫血、蛋白尿和进展性肾功能不全。血浆LCAT活性常减低或完全缺失。角膜混浊在儿童期即可出现,但肾功能受损多数出现在成人,常伴高血压。成熟HDL的缺乏将导致血浆内的脂蛋白与红细胞膜上的脂质交换障碍,引起过多的胆固醇在红细胞膜的蓄积,导致红细胞膜稳定性下降,造成溶血性贫血。LCAT缺乏症患者的死亡主要原因是肾脏受累和进展性肾功能不全,患者一般在4050岁进入终末期肾病(ESRD)。
LCAT造成肾脏损害的机制目前还不十分清楚。研究表明Lp-X可能参与了肾脏损害,Lp-X是一种异常的LDL ,富含磷脂和未酯化胆固醇,在FLD患者中水平明显升高。Gjone等[5]观察2位相同突变位点LCAT缺陷的亲属患者,不存在肾脏损害的患者血浆中未检测到Lp-X,而含有较高水平Lp-X的患者出现肾功能受损。Maryse等[6]在家系筛查中也发现类似的现象,推测Lp-X可能是造成肾脏损害的主要原因之一。Zhu等[15]利用LCAT酶缺乏的动物实验也证明小鼠体内Lp-X的含量升高,且与肾脏损害是相关的。当然,Lp-X并不是LCAT缺乏症患者所特异,因为一些胆管梗阻性疾病,Lp-X的水平也常常升高。
LCAT缺乏症的诊断及鉴别诊断NS或蛋白尿患者对激素治疗效果不佳,而且出现与NS不相符的血脂代谢异常,如HDL降低、游离胆固醇升高、VLDL升高、ApoA1降低,应当考虑是否存在LCAT缺乏症,尤其是HDL严重缺乏,具有较大的提示意义。但HDL的代谢涉及多种因素的参与,除LCAT基因突变(FED、FLD)外,另外一些遗传性疾病也能导致HDL缺乏和(或)成熟障碍,如ApoA1基因突变(家族性ApoA1缺陷综合征)[16],ABCA1基因突变(Tangier病)[17]。因此还需结合临床表现及其他血浆中的脂蛋白水平和成分进行综合分析,才能初步判断造成HDL减低的原因。
LCAT缺乏症、Fabry病[18]、Gaucher病[19]、Niemann-Pick病[20]和Ⅲ型高脂蛋白血症[21]均属脂质代谢异常性疾病,其发生机制、临床表现和病理改变的鉴别要点见表1。此外脂质沉积在肾小球内还可见于先天性肝内胆管发育不良征[22,23]。因此,肾小球内出现较多的泡沫细胞,一定要完善患者血脂方面的检查,分析血脂中各种成分的变化,找出突出的特点,结合光镜、免疫荧光及电镜的改变特点,初步判断患者的代谢异常的类型,结合酶活性的检测及相关基因的全段筛查,有助于疾病的确诊。
LCAT缺乏症的治疗目前 LCAT缺乏症尚无确切的治疗方法,一般采取对症治疗,包括降脂治疗、纠正贫血、降压及减少蛋白尿。透析能提高进入ESRD患者的生存率。Tsuchiya等[24]报道1例透析长达20余年的LCAT缺乏症患者,尽管存在腹主动脉钙化,但未发生心血管事件。进入ESRD患者也可选择肾移植,但移植肾会出现与自体肾相类似的病理改变,且不能改善全身脂质代谢异常[25-27]。复发对移植肾功能影响较小,移植肾存活较长。因此寻找治疗LCAT缺乏症的新方法显得十分重要。输注正常人新鲜冰冻血浆可能对LCAT患者有一定的疗效,但长期输注血浆可能会引起感染,且费用非常昂贵[28]。LCAT酶替代疗法有望成为治疗LCAT缺乏症患者的新方法[29,30]。
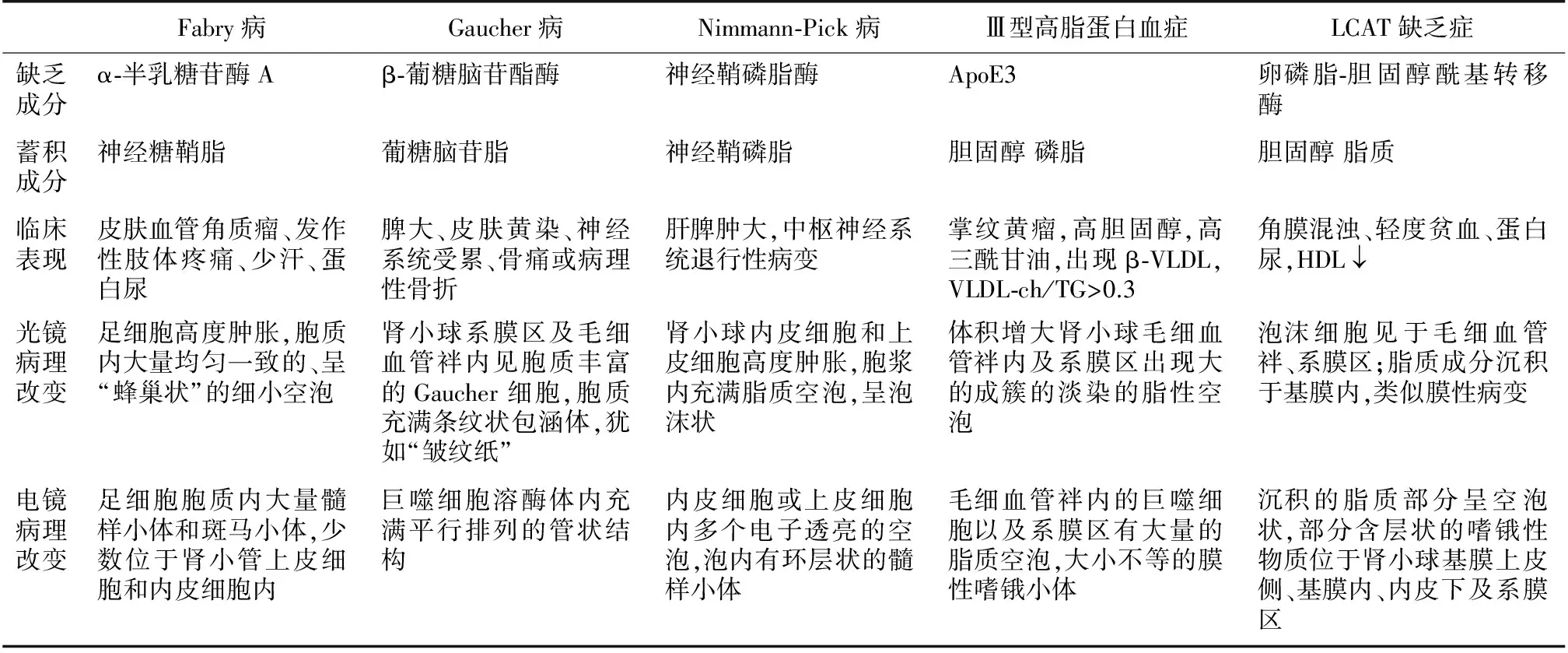
表1 脂质代谢异常沉积性疾病的鉴别诊断
小结:LCAT缺乏症是一类较罕见的常染色体隐性遗传性脂质代谢性疾病,是细胞外胆固醇代谢和转运的关键酶LCAT功能缺失所致。临床表现包括角膜混浊、贫血,肾脏受累表现为不同程度的蛋白尿和进展性肾功能不全。血浆LCAT活性常减低或完全缺失,血HDL水平下降。肾脏损害病理表现肾小球系膜区和GBM膜内大量脂性空泡及部分嗜锇性物质,免疫荧光多阴性,LCAT基因检测对诊断遗传性LCAT非常重要。
1Kuivenhoven JA,Pritchard H,Hill J,et al.The molecular pathology of lecithin:cholesterol acyltransferase (LCAT) deficiency syndromes.J Lipid Res,1997,38(2):191-205.
2Shoji K,Morita H,Ishigaki Y,et al.Lecithin-cholesterol acyltransferase (LCAT) deficiency without mutations in the coding sequence:a case report and literature review.Clin Nephrol,2011,76(4):323-328.
3Li M,Kuivenhoven JA,Ayyobi AF,et al.T→G or T→A mutation introduced in the branchpoint consensus sequence of intron 4 of lecithin:cholesterol acyltransferase (LCAT) gene:intron retention causing LCAT deficiency.Biochim Biophys Acta,1998,1391(2):256-264.
4Frascà GM,Soverini L,Tampieri E,et al.A 33-year-old man with nephrotic syndrome and lecithin-cholesterol acyltransferase (LCAT) deficiency.Description of two new mutations in the LCAT gene.Nephrol Dial Transplant,2004,19(6):1622-1624.
5Gjone E,Blomhoff JP,Skarbovik AJ.Possible association between an abnormal low density lipoprotein and nephropathy in lecithin:cholesterol acyltransferase deficiency.Clinica Chimica Acta,1974,54(1):11-8.
6Guerin M,Dachet C,Goulinet S,et al.Familial lecithin:cholesterol acyltransferase deficiency:molecular analysis of a compound heterozygote:LCAT (Arg→Trp) and LCAT (Tyr→Stop).Atherosclerosis,1997,131(1):85-95.
7Takahashi S,Hiromura K,Tsukida M,et al.Nephrotic syndrome caused by immune-mediated acquired LCAT deficiency.J Am Soc Nephrol,2013,24(8):1305-1312.
8Simonelli S,Gianazza E,Mombelli G,et al.Severe high-density lipoprotein deficiency associated with autoantibodies against lecithin:cholesterol acyltransferase in non-Hodgkin lymphoma.Arch Intern Med,2012,172(2):179-181.
9Vaziri ND,Liang K,Parks JS.Acquired lecithin-cholesterol acyltransferase deficiency in nephrotic syndrome.Am J Physiol Renal Physiol,2001,280(5):F823-828.
10 张小瑛,崔若兰.改良的 LCAT 测定及其在肾脏疾病中的临床意义.肾脏病与透析肾移植杂志,1995,4(4):318-319.
11 Vaziri ND,Liang K,Parks JS.Down-regulation of hepatic lecithin:cholesterol acyltransferase gene expression in chronic renal failure.Kidney Int,2001,59(6):2192-2196.
12 Valdemarsson S,Hedner P,Nilsson-Ehle P.Treatment of hyperthyroidism:effects on hepatic lipase,lipoprotein lipase,LCAT and plasma lipoproteins.Scand J Clin Lab Invest,1984,44(3):183-189.
13 Lager DJ,Rosenberg BF,Shapiro H,et al.Lecithin cholesterol acyltransferase deficiency:ultrastructural examination of sequential renal biopsies.Mod Pathol,1991,4(3):331-335.
14 Cirera S,Julve J,Ferrer I,et al.Molecular diagnosis of lecithin:cholesterol acyltransferase deficiency in a presymptomatic proband.Clin Chem Lab Med,1998,36(7):443-448.
15 Zhu X,Herzenberg AM,Eskandarian M,et al.A Nove in Vivo Lecithin-Cholesterol Acyltransferase (LCAT)-Deficient Mouse Expressing Predominantly LpX Is Associated with Spontaneous Glomerulopathy.Am J Pathol,2004,165(4):1269-1278.
16 Schaefer EJ,Heaton WH,Wetzel MG,et al.Plasma apolipoprotein A-1 absence associated with a marked reduction of high density lipoproteins and premature coronary artery disease.Arteriosclerosis,1982,2(1):16-26.
17 Brooks-Wilson A,Marcil M,Clee SM,et al.Mutations in ABC1 in Tangier disease and familial high-density lipoprotein deficiency.Nat genet,1999,22(4):336-345.
18 Alroy J,Sabnis S,Kopp JB..Renal pathology in Fabry disease.J Am Soc Nephrol,2002,13(suppl 2):S134-138.
19 Santoro D,Rosenbloom BE,Cohen AH.Gaucher disease with nephrotic syndrome:response to enzyme replacement therapy.Am J Kidney Dis,2002,40(1):E4.
20 陈惠萍,吴波.Niemann—Pick 病的肾脏损害.肾脏病与透析肾移植杂志,1998,7(1):88-91.
21 Balson KR,Niall JF,Best JD.Glomerular lipid deposition and proteinuria in a patient with familial dysbetalipoproteinaemia.J Intern Med,1996,240(3):157-159.
22 Habib R,Dommergues JP,Gubler MC,et al.Glomerular mesangiolipidosis in Alagille syndrome (arteriohepatic dysplasia).Pediatr Nephrol,1987,1(3):455-464.
23 Russo PA,Ellis D,Hashida Y.Renal histopathology in Alagille’s syndrome.Pediatr Pathol,1987,7(5-6):557-568.
24 Tsuchiya Y,Ubara Y,Hiramatsu R,et al.A case of familial lecithin-cholesterol acyltransferase deficiency on hemodialysis for over 20 years.Clin Nephrol,2011,76(6):492-498.
25 Panescu V,Grignon Y,Hestin D,et al.Recurrence of lecithin cholesterol acyltransferase deficiency after kidney transplantation.Nephrol Dial Transplant,1997,12(11):2430-2432.
26 Horina JH,Wirnsberger G,Horn S,et al.Long-term follow-up of a patient with lecithin cholesterol acyltransferase deficiency syndrome after kidney transplantation.Transplantation,1993,56(1):233-236.
27 Strøm EH,Sund S,Reier-Nilsen M,et al.Lecithin:cholesterol acyltransferase (LCAT) deficiency:renal lesions with early graft recurrence.Ultrastruct Pathol,2011,35(3):139-145.
28 Norum KR,Gjone E.The effect of plasma transfusion on the plasma cholesterol esters in patients with familial plasma lecithin:cholesterol acyltransferase deficiency.Scand J Clin Lab Invest.,1968,22(4):339-342.
29 Rousset X,Vaisman B,Auerbach B,et al.Effect of recombinant human lecithin cholesterol acyltransferase infusion on lipoprotein metabolism in mice.J Pharmacol Exp Ther,2010,335(1):140-148.
30 Stoekenbroek RM,van den Bergh Weerman MA,Hovingh GK,et al.Familial LCAT deficiency:from renal replacement to enzyme replacement.Neth J Med,2013,71(1):29-31.
