Toll样受体9在嘌呤霉素氨基核苷导致足细胞损伤中的作用
2014-03-21梁耀军鲍文朵娜周显光张明超刘志红施少林
夏 虹 梁耀军 何 静 鲍文朵娜 周显光 张明超 刘志红 施少林
Toll样受体(TLR)是一类识别病原体相关分子模式(PAMP)的受体家族,主要分布于免疫细胞,在人体由10个成员组成。TLR结合配体后,通过MyD88依赖途径或MyD88非依赖途径进行信号转导。经典的MyD88依赖途径主要通过TIR结构域的作用使MyD88活化并招募丝氨酸/苏氨酸蛋白激酶4(IRAK4),促使IRAK4介导的IRAK1发生磷酸化。IRAK1磷酸化后继续招募肿瘤坏死因子受体相关因子6(TRAF6)[1-3],TRAF6激活后诱导两种不同的信号通路:一方面最终激活核转录因子κB(NF-κB),导致一系列炎症因子的产生[如肿瘤坏死因子α(TNF-α)、白细胞介素12(IL-12)等[1,4,5]];另一方面通过旁路信号途径激活丝裂原活化蛋白激酶(MAPK)通路,主要有蛋白激酶(ERK)、c-Jun N端激酶(JNK)及p38 MAPK组成[6]。TLR9比较特殊,存在于内体膜上[7],其配体通过内吞作用进入细胞内,然后在内体与TLR9结合并激活TLR9信号通路,从而活化免疫细胞,发挥免疫学效应。病毒和细菌中的非甲基化CpG-DNA是TLR9的天然配体,近年来研究者发现,一些内源性物质也可通过TLR9发挥免疫活性作用,如氧化型DNA[8]、线粒体DNA(mtDNA)[9,10]、富含低甲基化CpG-DNA免疫复合物等[11-15]。
足细胞是重要的肾脏固有细胞,足细胞损伤引起的肾小球滤过屏障通透性改变和蛋白尿的产生被认为是肾小球硬化发生发展的中心始动因素[16]。研究表明,NF-κB在足细胞损伤产生蛋白尿的过程中发挥了重要的作用[17,18];p38 MAPK信号通路激活后参与足细胞损伤和凋亡[19,20]。近年的研究发现,TLR9在狼疮型肾病等疾病状态下的足细胞从无到有的表达[21],而TLR9能激活NF-κB和p38 MAPK,这提示TLR9可能参与了这些疾病的足细胞的损伤。
嘌呤霉素氨基核苷(PAN)可引起大鼠等实验动物出现肾病综合征,是经典的肾小球微小病变动物模型的造模药物。目前认为PAN可引起实验动物足细胞足突融合,造成非免疫因素介导的[22,23]、单纯足细胞病变[24]。以PAN处理体外培养的足细胞,可导致足细胞内NF-κB和p38 MAPK信号通路的激活[19,25],并引起足细胞骨架排列紊乱、凋亡等变化[26,27],因而是研究足细胞损伤机制的理想模型。
近期我们发现,PAN能上调体外培养的足细胞中TLR9的表达。鉴于TLR9信号能激活NF-κB和p38 MAPK,我们提出TLR9可能参与了PAN对足细胞的损伤作用。本研究利用若干分子生物学手段证明了该假说,从而揭示了足细胞损伤的一个新的分子机制。
材料与方法
实验动物6周龄Wistar雄性大鼠,体重140~160g,清洁级(南京军区南京总医院试验动物中心提供)。
材料
主要试剂永生化的人足细胞株(HPC)由英国Bristol大学Moin.A.Saleem教授惠赠;完全培养基为RPMI1640培养基含10%胎牛血清、青霉素、链霉素各100 U/ml(美国Gibco)及1% Insulin-Transferrin-Selenium(Invitrogen公司)。其他试剂包括18s rRNA、人的TLR9、IRAK1、TRAF6、IL-12等引物、细胞转染试剂Lipofectamine 2000及质粒DNA提取试剂盒(PureLink® HiPure Plasmid DNA Purification Kits)(均购自Invitrogen公司);总p65及p38 MAPK蛋白抗体、磷酸化p65及p38 MAPK抗体(美国Cell Signaling Technologies);TLR9和CD2AP抗体(Abcam);PAN和podocin抗体(Sigma);Alexa Fluor® 647 Annexin V和Propidium Iodide Solution(Biolegend); 逆转录试剂盒DRR037A和定量PCR试剂DRR820A(Takara);RIPA、BCA蛋白测定试剂盒(上海碧云天公司);抗兔二抗(Bioworld)、GAPDH抗体(Kangchen)、psiRNA-hTLR9(si-TLR9)质粒(Invivogen);RNA提取试剂盒(Ambion);E.coli DH5a 感受态细菌(北京全式金公司)。
方法
足细胞培养在5% CO2及饱和湿度条件下,足细胞先于33℃进行增殖,然后在37℃培养10~14d。处理细胞前先血清饥饿12h。以PBS和25~100 μg/ml的PAN刺激3h、6h、12h、24h或48h,然后收集细胞分别用于免疫印迹分析、定量逆转录PCR(qRT-PCR)基因表达检测和凋亡的流式细胞分析。
PAN肾病模型建立取6周龄雄性Wistar大鼠,体重140~160g,采用单次颈静脉注射法,将4 mg/100g体重剂量的PAN,从左侧颈静脉静脉注入,5 min内完成注射完毕。对照组动物同法注射相同剂量的生理盐水。实验设立对照组(n=8)和试验组(n=8),注射后20d处死大鼠。
Western Blot印迹检测25 cm2培养瓶中的细胞在处理完成后,以预冷的PBS液洗涤,加入150 μl预冷RIPA(Radio Immunopreciprtation Assay)裂解液(按1 ml裂解液加40 μl的25×蛋白酶抑制剂、100 μl的10×磷酸酶抑制剂),冰浴30 min,刮勺收集样本,4℃下11 500 r/min离心15 min,取上清,用BCA蛋白测定试剂盒测定蛋白浓度。细胞裂解液加入上样缓冲液后煮沸变性;样品经10%或8% SDS-聚丙烯酰胺凝胶电泳分离后,使用半干转移系统将蛋白转移至PVDF膜(电压10V,30~50 min)。将膜浸入含5%脱脂牛奶的TBST溶液(20 mmol/L Tris-HCl,pH 7.14,150 mmol/L NaCl,0.1% Tween20),室温孵育60 min;加入一抗(兔抗p65和抗磷酸化pp65,1∶1 000;兔抗p38和pp38,1∶1 000;鼠抗TLR9,1∶1 000;兔抗podocin,1∶1 000;兔抗CD2AP,1∶1 000),于4 ℃孵育过夜;经TBST洗涤3次(10 min/次)后,加入HRP标记的羊抗兔或羊抗鼠二抗(1∶15 000),室温孵育1h;经TBST洗涤后,用增强的化学发光系统(ECL,Millipore)显色,在GIS凝胶图像分析系统照相扫描蛋白条带的吸光度并分析处理。
qRT-PCR检测TLR9、IL-12的表达水平收集足细胞,总RNA按Ambion试剂盒方法提取并用Nanodrop测定纯度和浓度,再使用逆转录试剂盒(TakaRa,DRR037A)进行cDNA合成。qRT-PCR引物为人TLR9:5′-CCGTGACAATTACCTGGCCTTC-3′和5′-CAGGGCCTTCAGCTGGTTTC-3′。IL-12:5′-GGCCGTCAGCAACATGCTCCA-3′和5′-GGCACAGGGCCATCATAAAAGAGGT-3′。IRAK1:5′-CCACCCCAGTTCTATCATACTCC-3′和5′-GGCCAACAAGGTTACAGACTT-3′。TRAF6:5′-TTTGCTCTTATGGATTGTCCCC-3′和5′-CATTGATGCAGCACAGTTGTC-3′。参照物18s rRNA:5′-TTCTCGATTCCGTGGGTGG和5′-AGCATGCCAGAGTCTCGTTC。qPCR使用SYBR Green 法,qRT-PCR热循环条件:95℃ 30s预变性后,进行95℃ 5s、60℃ 30s的热循环,共40次;使用仪器为ABI 7900HT Fast Real time System。读取临界循环数(CT)进行记录分析。以正常对照细胞为矫正样本,用比较阈值法计算目的基因的相对含量(2-△△ct)。
流式细胞仪检测细胞凋亡采用膜联蛋白V/碘化丙啶(AnnexinV/PI)双染的流式细胞术检测PAN 诱导的足细胞凋亡。收集接种于六孔板的各组细胞,用PBS 洗2遍;按试剂盒(Biolegend)方法,用400 μl-1×Binding Buffer 悬浮细胞,浓度大约为1×106个/ml,在细胞悬浮液中加入Alexa Fluor 647 AnnexinV 和PI,轻轻混匀后室温避光条件下孵育10~15 min,在1h内用流式细胞仪(型号FACS ARIA,BD公司)检测细胞凋亡。
大鼠肾组织免疫组化染色取石蜡包埋的组织切片,经过二甲苯→二甲苯→无水乙醇→95%乙醇→75%乙醇进行脱蜡复水处理后,用蒸馏水清洗3遍,0.3%过氧化氢处理15 min,经微波处理后,用10%小牛血清孵育10 min,加入TLR9抗体(1∶50),4 ℃孵育过夜;经PBS洗涤5 min,加入二抗,室温孵育30 min,再进行DAB显色(约2 min,但以镜下观察为准);经苏木素复染3 min,用中性树胶(Sigma,St.Louis,MO)封片,置显微镜(Cannon,Japan)下观察。
TLR9干扰后对足细胞损伤的影响使用Lipofectamine 2000将si-TLR9和对照质粒psiRNA-h7SK(si-NC)转染足细胞(密度约80%),质粒转染按产品说明进行;转染24h后加入PAN处理24h,收集细胞分别用于qRT-PCR,Western Blot印迹,流式细胞分析等,以确定TLR9抑制对PAN损伤作用的影响。
统计学分析所有数据均为3次独立实验结果,以均数±标准差表示。采用SPSS 18.0统计软件进行统计学分析,两组之间比较采用t检验,P<0.05为差异有统计学意义,P<0.01为统计学差异显著。
结 果
PAN上调足细胞中TLR9信号通路相关分子的表达与正常对照组相比,经50 μg/ml PAN刺激的足细胞,其TLR9的mRNA水平显著增加(P<0.05)(图1A),蛋白表达也相应增加(图1B、C)。 此外,TLR9信号通路中的两个组分IRAK1和TRAF6的mRNA水平明显上调(图1D), 蛋白水平相应上调 (图1E、F)。
PAN上调NF-κB和p38 MAPK磷酸化水平以不同浓度PAN(0、25 μg/ml、50 μg/ml)处理足细胞24h, 与正常对照组相比,pp38随着PAN浓度增加而增加(图2A); 用50 μg/ml的PAN处理足细胞不同时间(0、6h、12h、24h)发现,6h时p65磷酸化就已经增加,并持续至24h(图2B)。同时,NF-κB信号通路下游的炎症因子IL-12的表达也发生了改变,即与对照组相比,PAN刺激组的IL-12 mRNA在12h和24h均显著上调(图2C)。
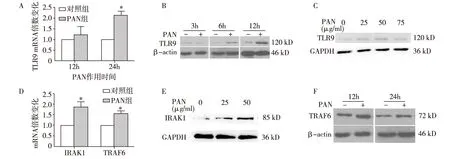
图1 PAN上调足细胞中TLR9信号通路相关分子的表达
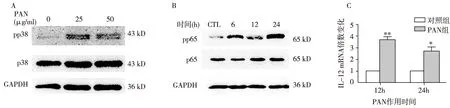
图2 PAN激活NF-κB和p38 MAPK信号通路
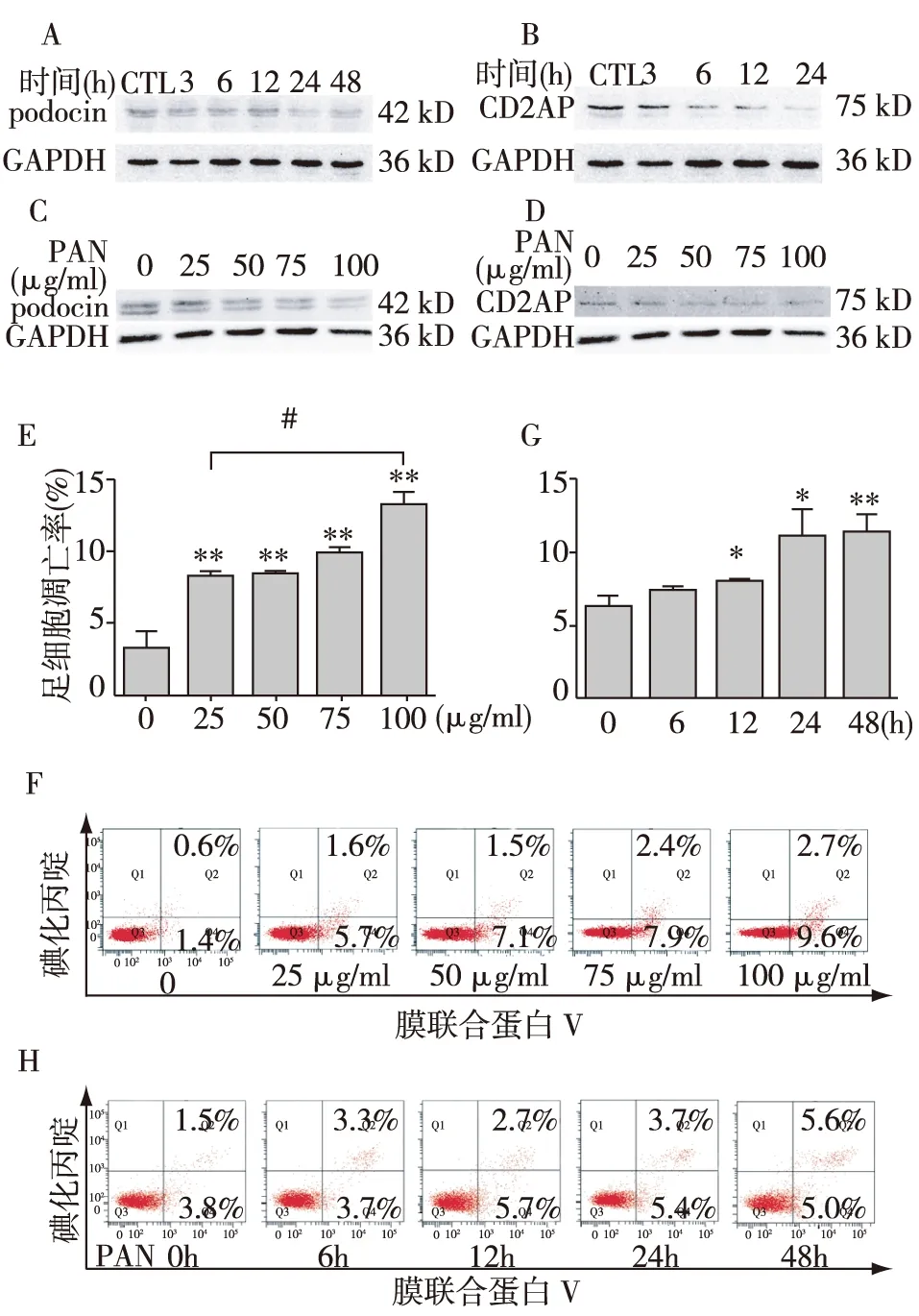
图3 PAN下调足细胞标记物的表达并增加细胞凋亡
PAN下调足细胞标记物并诱导足细胞凋亡以50 μg/ml PAN处理足细胞不同时间(0、3h、6h、12h、24h、48h),Western Blot印迹分析显示,足细胞标记物podocin和CD2AP从3h开始表达逐渐减低(图3A、3B)。此外,随PAN浓度增加(0、25 μg/ml、50 μg/ml、75 μg/ml、100 μg/ml)podocin和CD2AP蛋白水平呈逐渐降低趋势(图3C、D)。足细胞凋亡率随着PAN浓度增加而增加(图3E、F);以50 μg/ml PAN处理足细胞不同时间(0、6h、12h、24h),与正常对照组相比,25 μg/ml的PAN即可显著促进足细胞凋亡(P<0.01),而100 μg/ml的PAN能更显著提高足细胞的凋亡率(与25 μg/ml浓度相比,P<0.01; 图3E、F)。与正常对照组比较,PAN 处理足细胞6h的凋亡率为7.53%±0.29%,两者无统计学差异(P=0.059);而在PAN刺激12h时,约8.40%±0.23%的足细胞出现凋亡,与对照组存在统计学差异(P<0.05);在24h和48h时间点,凋亡率进一步提高增加(24h,P<0.05;48h,P<0.01),但48h与24h的凋亡率无明显变化(P=0.873)(图3G、H)。这些结果证明PAN能够诱导体外培养的足细胞发生损伤,是一个理想的足细胞损伤模型。
PAN上调大鼠足细胞TLR9的表达为了解TLR9信号通路是否参与PAN诱导的大鼠足细胞损伤,我们检测了TLR9在PAN大鼠足细胞的表达。免疫组化结果表明,与正常对照组相比,PAN组大鼠肾小球中的TLR9染色强度明显增高(图4A)。我们用筛网过滤法得到PAN大鼠和正常大鼠的肾小球,提取总RNA后,进行qRT-PCR分析显示,与正常对照组相比,PAN大鼠肾小球中TLR9的mRNA水平显著增加(P<0.05)(图4B)。这提示TLR9信号通路的激活可能在多种肾损伤模型中存在,而且提示PAN大鼠可能是研究TLR9在足细胞损伤中作用的合适活体模型。
足细胞TLR9干扰能减弱PAN活化NF-κB和p38及上调IL-12表达的效应将足细胞转染TLR9干扰psiRNA-hTLR9质粒及对照质粒,qRT-PCR结果显示,TLR9干扰组细胞(si-TLR9)中TLR9 mRNA 水平较对照组(si-NC)明显降低(P<0.05,图5A)。此外,足细胞转染质粒后,给予或不给予PAN(50 μg/ml)刺激,再提取蛋白进行TLR9免疫印迹分析发现,PAN增加si-NC组细胞(si-NC+PAN)TLR9表达,但PAN处理si-TLR9组细胞(si-TLR9+PAN)时,TLR9的增加幅度明显小于si-NC+PAN组(图5B)。 以上结果表明TLR9干扰质粒成功减低了TLR9 mRNA和蛋白的表达。
我们对上述细胞进行了NF-κB和p38的活性的检测。si-TLR9+PAN组与si-NC+PAN组相比,p38磷酸化水平和NF-κB p65磷酸化水平均明显降低(图5C、D)。因此,在足细胞PAN损伤模型中,TLR9干扰能抑制p38和NF-κB信号通路。为进一步证明,我们检测下游炎症因子IL-12的表达发现,PAN显著上调si-NC+PAN组细胞的IL-12 mRNA(与si-NC组相比,P<0.05,图5E);而TLR9干扰能显著减弱PAN对IL-12 mRNA的诱导作用(si-NC+PAN组与si-TLR9+PAN组相比,P<0.05,图5E)。
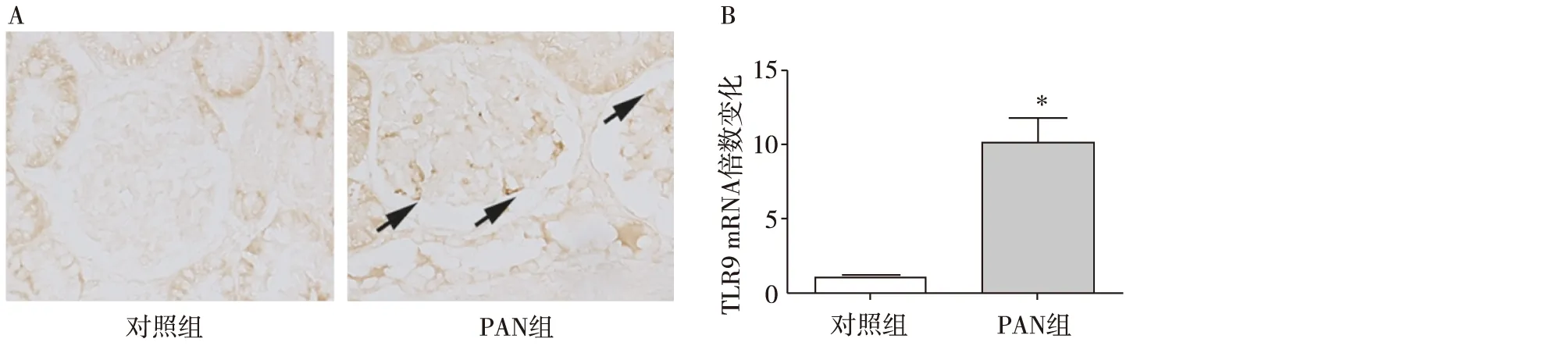
图4 PAN诱导上调大鼠肾组织TLR9表达

图5 足细胞TLR9干扰对NF-κB和p38磷酸化水平及IL-12表达的影响
TLR9干扰能减轻PAN诱导的足细胞凋亡将足细胞转染TLR9干扰质粒及对照质粒并经PAN(50 mg/ml)处理24 h 后,进行Annexin V染色和流式细胞分析发现,PAN显著增加si-NC+PAN组细胞的凋亡 (si-NC+PAN与si-NC组相比,P<0.01,图6A);而TLR9干扰能消除PAN诱导的细胞凋亡,即与si-NC+PAN组相比,si-TLR9+PAN组足细胞凋亡率明显减低(P<0.01), 且与si-NC及si-TLR9组细胞的凋亡率无显著差别(图6A)。代表性的流式细胞分析结果见图6B。

图6 TLR9干扰质粒转染对足细胞凋亡的影响
讨 论
足细胞位于肾小球基膜(GMB)外侧,相邻的足细胞之间通过裂隙膜紧密相连,构成肾小球滤过的关键屏障[28]。足细胞功能的降低、损伤及丢失是导致各种肾小球疾病发生发展的重要原因之一[29]。研究足细胞对损伤刺激的应答,寻找保护足细胞的新途径对于治疗肾脏疾病或者延缓肾脏病进展至关重要。在这类研究中,通常需要借助各种损伤模型,包括若干药物诱导足细胞损伤的模型,研究足细胞损伤的分子机制,从而确定保护或治疗足细胞的分子靶点。
PAN诱导的肾脏损伤模型被认为能模拟微小病变(MCD)和局灶节段性肾小球硬化(FSGS),并以足细胞损伤为其突出特征,常表现为足突融合、足细胞凋亡、足细胞减少等。已有研究证明PAN造成的细胞内氧化应激是足细胞损伤产生蛋白尿的重要因素[30,31]。给予氧自由基清除剂预处理后,同样PAN刺激,足细胞的受损程度得到部分缓解[31,32]。Marshall等[33]发现,PAN介导产生的活性氧能够直接导致 DNA损伤,上调了特异性的细胞周期检验点,最终导致细胞周期停滞。此外,还有研究证实PAN刺激足细胞时能激活NF-κB和p38 MAPK信号通路[25,34],足细胞内NF-κB上调能导致肾小球肾炎和蛋白尿[18],而抑制肾小球肾炎模型足细胞的NF-κB 信号通路,能减轻足细胞损伤和蛋白尿[17]。足细胞内p38 MAPK激活与细胞骨架完整性改变和凋亡密切相关[19,20],p38 MAPK的抑制剂能有效防止足细胞标志物的减少和蛋白尿发生。这些研究提示NF-κB和p38 MAPK信号可能介导PAN诱导的足细胞损伤。
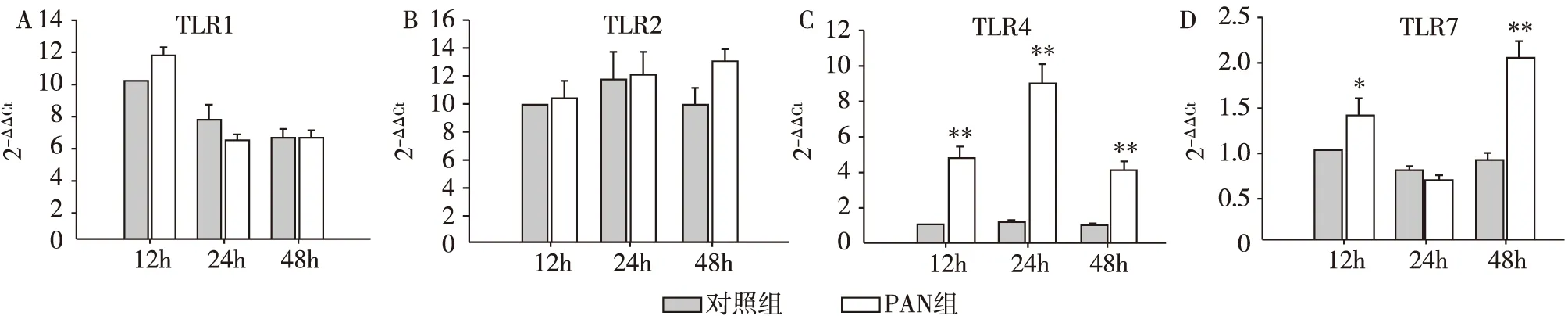
图7 足细胞中TLR1、TLR2、TLR4和TLR7的表达分析
众所周知,TLR信号在哺乳动物的固有或适应性免疫反应中发挥着重要作用,是联系先天免疫和后天免疫的桥梁。免疫细胞中TLR9信号通路激活通常可引起下游NF-κB 和p38 MAPK信号通路活化,导致细胞产生炎症因子。与活体足细胞不同,已有研究证明体外培养的足细胞表达TLR9[35]。本研究也证实了这一结果,而且我们还发现,PAN处理足细胞能上调TLR9的表达,且成时间和浓度依赖性;用PAN处理足细胞发现,与对照组相比PAN组的NF-κB和p38 MAPK信号得以激活,p65和p38的蛋白磷酸化水平明显上调,这些结果均提示除了氧化应激,TLR9可能是PAN导致足细胞损伤的又一机制。为了证明TLR9信号参与了PAN诱导的足细胞损伤,我们利用RNA干扰技术降低足细胞TLR9表达发现,TLR9抑制可降低PAN引起的NF-κB 和p38磷酸化、并减少足细胞凋亡,从而验证了我们的假说。我们也进行了足细胞TLR9过表达,发现NF-κB 和p38磷酸化及足细胞凋亡并不能进一步增强,这可能提示我们所使用的足细胞系,其TLR9含量已达饱和,使配体能被充分利用,因而外源TLR9的过表达并不能增强TLR9信号。
上文中已经提到PAN处理足细胞能上调TLR9的表达,并成时间和浓度依赖性,但具体的机制尚不清楚。研究表明在其他的疾病中(例如动脉粥样硬化),内皮细胞中氧化应激的产物氧化低密度脂蛋白能够通过诱导自噬和mtDNA损伤最终增加TLR9的表达[36,37]。目前我们已经知道PAN能够造成足细胞发生氧化应激,可能类似的机制也存在于足细胞中,最终导致足细胞内TLR9的上调[38]。
免疫细胞中除了TLR9,其他TLRs分子激活后同样能通过MyD88依赖途径或MyD88非依赖途径引起下游NF-κB 和p38 MAPK信号通路活化。近年来研究表明体外培养的足细胞也不同程度地表达TLRs,比如TLR1~6[35]、TLR10;此外,PAN处理足细胞可导致TLR2~6的表达不同程度地增加,并能够诱导NF-κB发生核转移[25]。本研究证实除了TLR9,足细胞还组成性表达TLR1、TLR2、TLR4和TLR7,予PAN处理后TLR4和TLR7的mRNA水平显著增加(图7),因而TLR4和TLR7也可能参与了NF-κB 和p38 MAPK的激活。为确定TLR9的作用,我们进行了TLR9干扰,发现PAN引起的NF-κB 和p38磷酸化和细胞凋亡能被显著减轻,证明TLR9确实参与了PAN对足细胞的损伤作用。
在PAN处理的足细胞中,我们注意到TLR9下游的分子变化似乎早于TLR9的上调。可能的原因是,本研究中所使用的足细胞系均能检测到TLR9 mRNA及蛋白的表达,因而PAN的处理可能立刻产生TLR9配体,并与已经存在的TLR9结合并激活其信号通路,引起下游分子的变化。后续由PAN诱导的TLR9应该能进一步增强该信号和下游分子的变化。目前,我们猜测PAN能损伤线粒体,引起线粒体的自噬,从而使mtDNA(TLR9的可能配体)被带入内体/溶酶体,结合并激活其中的TLR9。
本研究中我们观察到PAN处理足细胞能够引起TLR9激活这一现象,但与TLR9相互作用的配体仍不明确。TLR9的天然配体是病毒和细菌中的非甲基化CpG-DNA,但新近研究发现TLR9也能够识别个体自身mtDNA。研究表明,在创伤或出血性休克情况下,损伤组织释放的mtDNA能够通过与TLR9相互作用刺激中性粒细胞并激活固有免疫反应[39]。除了细胞外mtDNA,有研究认为细胞内的mtDNA也可作用于TLR9的配体,比如逃避了自噬作用的mtDNA可通过TLR9参与心肌细胞的炎症反应并促进心肌炎及扩张型心肌病的发生和发展[10]。线粒体来源于细菌,mtDNA与细菌DNA具有相似性,含有非甲基化CpG基序。这些研究提示在足细胞PAN损伤模型中,PAN可能损伤线粒体并释放mtDNA,该mtDNA可能成为TLR9配体,激活TLR9信号,促进足细胞损伤。不过该假说有待于实验进一步证明。
有研究表明TLR9在正常小鼠足细胞中不表达[40]。Machida等[21]也证明正常人肾脏固有细胞同样不表达TLR9,但幼年狼疮患者足细胞存在TLR9高表达,并且TLR9也表达于其他足细胞病的部分患者,如MCD和FSGS等。此外,Batsford等[41]发现在其他疾病中,TLR9表达于肾小球固有细胞,比如多瘤病毒感染和溶血尿毒综合征。这些研究提示TLR9可能参与足细胞损伤过程。由于我们发现TLR9在的PAN足细胞损伤模型表达上调,因此,PAN的足细胞损伤模型可能是研究TLR9是否参与足细胞损伤的理想工具。本研究结果证明,TLR9通过NF-κB和p38 MAPK信号通路,介导了PAN诱导的足细胞损伤。提示TLR9在多种肾脏疾病中的足细胞表达可能促进了足细胞损伤,因而TLR9可能是这些肾脏病治疗的潜在靶点。
1Akira S,Takeda K,Kaisho T.Toll-like receptors:critical proteins linking innate and acquired immunity.Nat Immunol,2001,2(8):675-680.
2Häcker H,Vabulas R M,Takeuchi O,et al.Immune cell activation by bacterial CpG-DNA through myeloid differentiation marker 88 and tumor necrosis factor receptor—associated factor(TRAF) 6.J Exp Med,2000,192(4):595-600.
3Hemmi H,Takeuchi O,Kawai T,et al.A Toll-like receptor recognizes bacterial DNA.Nature,2000,408(6813):740-745.
4Anders HJ.A Toll for lupus.Lupus,2005,14(6):417-422.
5Heeg K and Zimmermann S.CpG DNA as a Th1 trigger.Int Arch Allergy Immunol,2009,121(2):87-97.
6Johnson GL,Lapadat R.Mitogen-activated protein kinase pathways mediated by ERK,JNK,and p38 protein kinases.Science,2002,298(5600):1911-1912.
7Latz E,Schoenemeyer A,Visintin A,et al.TLR9 signals after translocating from the ER to CpG DNA in the lysosome.Nat Immunol,2004,5(2):190-198.
8Ermakov AV,Konkova MS,Kostyuk SV,et al.Oxidized extracellular DNA as a stress signal in human cells.Oxid Med Cell Longev,2013,2013:649747.
9Zhang Q,Raoof M,Chen Y,et al.Circulating mitochondrial DAMPs cause inflammatory responses to injury.Nature,2010,464(7285):104-107.
10 Oka T,Hikoso S,Yamaguchi O,et al.Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure.Nature,2012,485(7397):251-255.
11 Lövgren T,Eloranta M L,Båve U,et al.Induction of interferon—α production in plasmacytoid dendritic cells by immune complexes containing nucleic acid released by necrotic or late apoptotic cells and lupus IgG.Arthritis Rheum,2004,50(6):1861-1872.
12 Viglianti GA,Lau CM,Hanley TM,et al.Activation of autoreactive B cells by CpG dsDNA.Immunity,2003,19(6):837-847.
13 Leadbetter EA,Rifkin IR,Hohlbaum AM,et al.Chromatin—IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors.Nature,2002,416(6881):603-607.
14 Barrat FJ,Meeker T,Gregorio J,et al.Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus.J Exp Med,2005,202(8):1131-1139.
15 Boulé MW,Broughton C,Mackay F,et al.Toll-like receptor 9-dependent and -independent dendritic cell activation by chromatin-immunoglobulin G complexes.J Exp Med,2004,199(12):1631-1640.
16 Kim YH,Goyal M,Kurnit D,et al.Podocyte depletion and glomerulosclerosis have a direct relationship in the PAN-treated rat.Kidney Int,2001,60(3):957-968.
17 Brähler S,Ising C,Hagmann H,et al.Intrinsic proinflammatory signaling in podocytes contributes to podocyte damage and prolonged proteinuria.Am J Physiol Renal Physiol,2012,303(10):F1473-F1485.
18 Hussain S,Romio L,Saleem M,et al.Nephrin deficiency activates NF-κB and promotes glomerular injury.J Am Soc Nephrol,2009,20(8):1733-1743.
19 Koshikawa M,Mukoyama M,Mori K,et al.Role of p38 mitogen-activated protein kinase activation in podocyte injury and proteinuria in experimental nephrotic syndrome.J Am Soc Nephrol,2005,16(9):2690-2701.
20 Schiffer M,Bitzer M,Roberts IS,et al.Apoptosis in podocytes induced by TGF-β and Smad7.J Clin Invest,2001,108(6):807-816.
21 Machida H,Ito S,Hirose T,et al.Expression of Toll-like receptor 9 in renal podocytes in childhood-onset active and inactive lupus nephritis.Nephrol Dial Transplant,2010,25(8):2430-2537.
22 Sanwal V,Pandya M,Bhaskaran M,et al.Puromycin aminonucleoside induces glomerular epithelial cell apoptosis.Exp Mol Pathol,2001,70(1):54-64.
23 Hagiwara M,Yamagata K,Capaldi R,et al.Mitochondrial dysfunction in focal segmental glomerulosclerosis of puromycin aminonucleoside nephrosis.Kidney Int,2006,69(7):1146-1152.
24 Pippin J W,Brinkkoetter P T,Cormack-Aboud F C,et al.Inducible rodent models of acquired podocyte diseases.Am J Physiol Renal Physiol,2009,296(2):F213-F229.
25 Srivastava T,Sharma M,Yew KH,et al.LPS and PAN-induced podocyte injury in an in vitro model of minimal change disease:changes in TLR profile.J Cell Commun Signal,2013,7(1):49-60.
26 Adams J L,Badger AM,Kumar S,et al.p38 MAP kinase:molecular target for the inhibition of pro-inflammatory cytokines.Prog Med Chem,2001,38:1-60.
27 Kimura C,Zhao QL,Kondo T,et al.Mechanism of UV-induced apoptosis in human leukemia cells:roles of Ca2+/Mg(2+)-dependent endonuclease,caspase-3,and stress-activated protein kinases.Exp Cell Res,1998,239(2):411-422.
28 Mundel P,Shankland SJ.Podocyte biology and response to injury.J Am Soc Nephrol,2002,13(12):3005-3015.
29 Wiggins RC.The spectrum of podocytopathies:a unifying view of glomerular diseases.Kidney Int,2007,71(12):1205-1214.
30 Beaman M,Birtwistle R,Howie AJ,et al.The role of superoxide anion and hydrogen peroxide in glomerular injury induced by puromycin aminonucleoside in rats.Clin Sci(Lond),1987,73(3):329-332.
31 Diamond JR,Bonventre JV,Karnovsky MJ.A role for oxygen free radicals in aminonucleoside nephrosis.Kidney Int,1986,29(2):478-483.
32 Thakur V,Walker PD,Shah SV.Evidence suggesting a role for hydroxyl radical in puromycin aminonucleoside-induced proteinuria.Kidney Int,1988,34(4):494-499.
33 Marshall CB,Pippin JW,Krofft RD,et al.Puromycin aminonucleoside induces oxidant-dependent DNA damage in podocytes in vitro and in vivo.Kidney Int,2006,70(11):1962-1973.
34 Zheng CX,Chen ZH,Zeng CH,et al.Triptolide protects podocytes from puromycin aminonucleoside induced injury in vivo and in vitro.Kidney Int,2008,74(5):596-612.
35 Shimada M,Ishimoto T,Lee PY,et al.Toll-like receptor 3 ligands induce CD80 expression in human podocytes via an NF-κB-dependent pathway.Nephrol Dial Transplant,2012,27(1):81-89.
36 Ding Z,Liu S,Wang X,et al.Oxidant stress in mitochondrial DNA damage,autophagy and inflammation in atherosclerosis.Sci Rep,2013,3:1077.
37 Ding Z,Liu S,Wang X,et al.LOX-1,Oxidant stress,mtDNA damage,autophagy,and immune response in atherosclerosis.Canadian Journal of physiology and Pharmacology,2014,92(999):1-7.
38 Rincon J,Romero M,Viera N,et al.Increased oxidative stress and apoptosis in acute puromycin aminonucleoside nephrosis.Int J Exp Pathol,2004,85(1):25-33.
39 Zhang Q,Itagaki K,Hauser C J.Mitochondrial DNA is released by shock and activates neutrophils via p38 map kinase.Shock,2010,34(1):55-59.
40 Anders HJ,Vielhauer V,Eis V,et al.Activation of toll-like receptor-9 induces progression of renal disease in MRL-Fas(lpr) mice.FASEB J,2004,18(3):534-536.
41 Batsford S,Duermueller U,Seemayer C,et al.Protein level expression of Toll-like receptors 2,4 and 9 in renal disease.Nephrol Dial Transplant,2011,26(4):1413-1416.
