钙蛋白酶通过降解突触蛋白Ⅰ介导脑出血后继发性神经元损伤
2014-03-10张振涛张国新王洪财熊婧胡丹张兆辉
张振涛,张国新,王洪财,熊婧,胡丹,张兆辉
脑出血后血肿形成后除占位效应损伤周围脑组织外,还会通过释放生物毒性物质,产生继发性神经损伤。脑出血继发性神经损伤的机制尚未完全阐明,但目前认为蛋白酶活化、红细胞破损造成局部铁超载等因素均参与神经损伤过程[1-2]。钙蛋白酶是体内一类重要的蛋白酶,在中枢神经系统中含量丰富。正常情况下,钙蛋白酶能够促进突触功能,参与记忆形成。但在疾病状态下由于神经元钙超载,导致钙蛋白酶过度活化。过度活化的钙蛋白酶通过降解其底物导致神经损伤。钙蛋白酶可降解微管相关蛋白,导致细胞骨架损伤[3];钙蛋白酶也可降解代谢型谷氨酸受体,使其失去正常功能[4]。另外,钙蛋白酶还可将P35降解为P25,介导神经损伤[5]。但迄今为止,钙蛋白酶在血肿周围脑组织继发性神经损伤的作用尚不明确。本研究利用在体和离体脑出血模型观察了钙蛋白酶通过降解其底物突触蛋白Ⅰ参与神经损伤的作用及其机制,以期为脑出血后继发性神经损伤寻找新的治疗靶点。
1 材料与方法
1.1 主要仪器及试剂 钙蛋白酶抗体和MDL28170购自Calbiochem公司,神经生长因子购自Sigma公司,钙蛋白酶抑制剂ALLN购自Santa Cruz公司,钙蛋白酶活性检测试剂盒购自Abcam公司。立体定向仪购自西北光学仪器厂,大鼠嗜铬细胞瘤细胞系(PC12细胞)由武汉大学典型培养物储藏中心提供。重组突触蛋白Ⅰ购自Novus公司,活化的calpain购自Abcam公司,突触蛋白Ⅰ抗体购自Cell Signaling公司。
1.2 自体血注射制作 在体脑出血模型10周龄雄性C57BL/6J小鼠购自武汉大学实验动物中心。实验动物随机分为4组:0 h组(对照组)、2 h组、12 h组和24 h组,每组10只小鼠。根据文献报道的方法制作自体血注射脑出血模型[6]。苯巴比妥(75 mg/kg)麻醉,局部剃毛、消毒。囟门前0.2 mm,侧开2 mm用牙科钻钻开颅骨,使用Hamilton注射器向脑内注射5 μl自体尾静脉血,速度为2 μl/min。停针5 min后继续进针0.7 mm,注射25 μl自体尾静脉血,停针10 min,缓慢拔针,用骨蜡封闭颅骨破损。分别于术后2 h、12 h、24 h开颅骨,提取血肿周围脑组织。0 h组在注射25 μl自体尾静脉血后立即取注射点周围脑组织。
1.3 钙蛋白酶对重组突触蛋白Ⅰ的降解 在试管内将10 μg纯化的重组突触蛋白Ⅰ(购自Novus公司)溶于钙蛋白酶缓冲液[50 mmol/L Tris-HCl,pH 7.5;100 mmol/L NaCl;2 mmol/L二硫苏糖醇(dithiothreitol,DTT)1 mmol/L乙二胺四乙酸二钠(edetate disodium,EDTA);3 mmol/L CaCl2],然后加入钙蛋白酶(购自Abcam公司),终浓度为5 U/ml。37℃孵育10 min、20 min、30 min后免疫印迹法检测突触蛋白Ⅰ的降解,以0 h的为对照组,设定此时蛋白浓度为100%。
1.4 PC12细胞培养及诱导分化 PC12细胞置于达尔伯克必需基本培养基(Dulbecco minimum essential medium,DMEM)中,内含10%(体积分数)热灭活小牛血清、青霉素100 U/ml、链霉素100 U/ml,于5%(体积分数)CO2,37℃培养箱内进行培养,隔天换液。药物处理前l周用神经生长因子(nerve growth factor,NGF)(终浓度为0.1 μg/ml)诱导PC12细胞神经元样分化。
1.5 3-(4,5-二甲基噻唑-2)-2,5-二苯基四氮唑溴盐染色法检测细胞活力 调节PC12细胞密度约2×105/ml,以100 μl/孔接种于96孔板,分别加入5 U/ml的钙蛋白酶或钙蛋白酶抑制剂处理2 h、4 h、8 h和12 h,每种处理设立6个复孔,每孔加5 mg/ml 3-(4,5-二甲基噻唑-2)-2,5-二苯基四氮唑溴盐[(3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide,MTT)]10 μl,继续培养4 h。弃培养基,每孔加二甲基亚砜100 μl,37℃孵育20 min,至颗粒溶解,在酶标仪上测定570 nm的吸光度值,将其作为反映PC12细胞活性和代谢状况的参数。细胞存活率=试验组吸光度值/对照组吸光度值×100%。
1.6 免疫印迹实验 收集小鼠脑组织,加细胞裂解液后在冰上裂解,1300 rpm离心10 min,取上清液,测定蛋白浓度后聚丙烯酰胺凝胶电泳,电转至硝酸纤维素膜上,然后用钙蛋白酶抗体4℃孵育过夜,二抗孵育后显色、曝光。
1.7 钙蛋白酶活性检测 根据钙蛋白酶活性检测试剂盒说明书,PC12细胞处理后使用裂解液裂解细胞,离心取上清液,蛋白定量后取100 μg蛋白,加入10 μl 10×反应缓冲液和5 μl钙蛋白酶底物,37℃避光反应1 h,然后使用400 nm激发波长、505 nm发射波长检测荧光强度。
1.8 统计学分析 采用Excel软件建立数据库,SPSS 17.0软件进行统计学分析。计量资料如符合正态分布采用“均数±标准差”表示;不符合正态分布,采用中位数、四分位数间距表示。多组间比较采用单因素方差分析(ANOVA),两两比较采用SNK-q检验。P<0.05为差异有显著性。
2 结果
2.1 血肿周围脑组织钙蛋白酶活性变化及突触蛋白Ⅰ降解 在颅内注射自体血后0 h、2 h、12 h、24 h收集血肿周围脑组织,检测钙蛋白酶活性,0 h、2 h、12 h、24 h蛋白酶活性读数分别为234.32、343.87、425.29和597.36。12 h组和24 h组与0 h组比较差异具有显著性(P<0.05)。使用突触蛋白Ⅰ抗体进行免疫印迹检查发现,血肿周围脑组织突触蛋白Ⅰ水平在血肿形成后逐渐下降,至血肿形成后24 h下降到0 h组的67%(F=8.056,P<0.01),提示出现突触蛋白Ⅰ降解(图1)。
2.2 钙蛋白酶对纯化的突触蛋白Ⅰ的降解 体外纯化的突触蛋白Ⅰ与5 U/ml蛋白酶在37℃孵育10 min、20 min、30 min后免疫印迹法检测突触蛋白Ⅰ表达,发现随着孵育时间的延长,突触蛋白Ⅰ水平逐渐下降,提示突触蛋白Ⅰ被钙蛋白酶降解。孵育10 min、20 min、30 min后突触蛋白Ⅰ含量分别下降至对照组(0 h组)的76.25%(P=0.022)、64.75%(P=0.002)和57.75%(P=0.001)。在钙蛋白酶抑制剂ALLN(50 μmol/L)和MDL28170(10 μmol/L)存在的情况下,孵育30 min后突触蛋白Ⅰ分别下降至对照组的87.00%和84.75%,与单纯钙蛋白酶处理组30 min组相比差异具有显著性(P<0.05)。该结果提示钙蛋白酶抑制剂能够抑制钙蛋白酶对突触蛋白Ⅰ的降解(图2)。
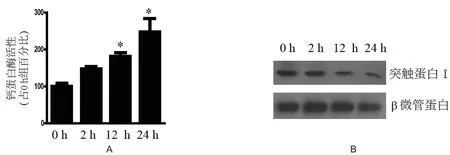
图1 血肿形成后不同时间点钙蛋白酶活性和突触蛋白Ⅰ表达变化注:A:钙蛋白酶活性变化;B:突触蛋白Ⅰ表达水平变化。*:与对照组比较,P<0.05
2.3 钙蛋白酶对神经元样分化的PC12细胞的损伤作用 PC12细胞经NGF处理后出现神经元样分化,加入外源性钙蛋白酶5 U/ml后PC12细胞活力逐渐下降,处理4 h、8 h和12 h后细胞活力分别下降到对照组的76%(P=0.002)、69.5%(P<0.001)和58.25%(P<0.01)。同时突触蛋白Ⅰ水平逐渐降低。钙蛋白酶抑制剂ALLN(50 μmol/L)处理组细胞活力升高到对照组的83%,而MDL28170(10 μmol/L)处理组细胞活力也升高到对照组的80.25%(与单纯钙蛋白酶处理12 h组相比,P<0.01),提示钙蛋白酶抑制剂具有保护作用(图3)。
3 讨论
脑出血后血肿周围神经元继发性损伤的分子机制尚未阐明,因此缺乏相应的治疗方法[7]。既往研究发现,脑出血后血肿释放一系列神经损伤因子,主要包括蛋白酶和铁离子,这些损伤因子导致血肿周围神经元损伤[8]。脑出血后神经元钙超载,可激活一系列钙离子依赖的神经损伤通路[9]。钙蛋白酶是一种重要的钙离子依赖的蛋白酶,推测其可能在脑出血后继发性神经损伤中发挥作用。但迄今为止钙蛋白酶在脑出血后继发性神经损伤中的作用目前尚不明确[10]。本研究使用自体静脉血注射制作大鼠脑出血模型,大鼠注射自体静脉血后均可见到血肿形成,与既往报道吻合[6]。本研究发现血肿形成后周围脑组织钙蛋白酶活性增高,并通过降解突触相关蛋白突触蛋白Ⅰ促进神经元损伤。
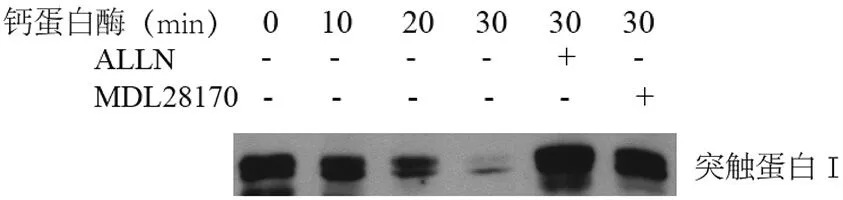
图2 活化的钙蛋白酶降解纯化的重组突触蛋白Ⅰ注:重组突触蛋白Ⅰ在试管内与纯化的钙蛋白酶共同孵育,可见随着孵育时间的延长,突触蛋白Ⅰ逐渐被降解。钙蛋白酶抑制剂ALLN和MDL28170能够抑制钙蛋白酶介导的突触蛋白Ⅰ降解
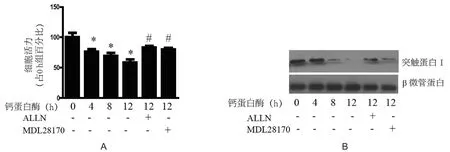
图3 钙蛋白酶及其抑制剂对细胞活力和突触蛋白Ⅰ表达的影响注:A:钙蛋白酶及其抑制剂对细胞活力的影响;B:钙蛋白酶及其抑制剂对突触蛋白Ⅰ表达的影响。*:与对照组(0 h组)比较,P<0.05;#:与12 h组比较,P<0.05
钙蛋白酶是一种钙离子依赖的蛋白酶,在体内参与一系列病理生理过程,包括细胞周期调控、细胞存活等[11-12],参与神经元长时程增强、血管内径调节等,因此在中枢神经系统中发挥重要的作用[13]。既往研究发现,在脑出血发生过程中,血肿周围组织会出现钙超载[9],我们推测本研究中观察到的钙蛋白酶活性增高是由神经元钙超载引起的。钙离子可直接结合到钙蛋白酶的催化中心将其活化[14],活化的钙蛋白酶可通过降解其底物发挥作用。
突触蛋白Ⅰ是一种突触前蛋白,可通过控制突触囊泡的释放调节突触功能。全长的突触蛋白Ⅰ包含5个结构域,在神经元电静息状态下,突触蛋白Ⅰ存在于突触囊泡中,维持突触囊泡的完整性,抑制神经递质的释放。在神经元动作电位的影响下,突触蛋白Ⅰ被磷酸化,与突触囊泡脱离接触,突触囊泡与突触前膜融合,将神经递质释放到细胞外,完成突触传递,然后去磷酸化的突触蛋白Ⅰ再次募集突触囊泡,为下一次动作电位做准备[15]。突触蛋白基因敲除小鼠出现突触囊泡分布异常、癫痫等病理表现,可见突触蛋白Ⅰ对突触正常功能是必不可少的[16]。
本研究观察到脑出血周围组织钙蛋白酶活性增高,同时突触蛋白Ⅰ表达水平逐渐降低,两者呈负相关。结合既往研究发现钙蛋白酶可降解突触相关蛋白[17],提示脑出血后活化的钙蛋白酶可能参与突触蛋白Ⅰ的降解。为了进一步明确钙蛋白酶和突触蛋白Ⅰ的直接相互作用,本研究在体外共孵育活化的钙蛋白酶和纯化的重组突触蛋白Ⅰ不同时间,以0 h组为对照组,此时钙蛋白酶尚未发挥作用。发现活化的钙蛋白酶可降解突触蛋白Ⅰ,并且该作用可被钙蛋白酶特异性抑制剂MDL28170[18]所阻断。该结果提示钙蛋白酶可直接降解突触蛋白Ⅰ,而不需要其他蛋白酶的参与。既往研究也发现钙蛋白酶能够降低突触蛋白Ⅰ的半衰期,而钙蛋白酶抑制剂能够阻断突触蛋白Ⅰ的降解,进一步支持本研究的结果[17]。
突触蛋白Ⅰ是一种突触前蛋白,控制突触囊泡的形成和神经递质的释放,在突触传递中起到关键作用[19],而且,正常的神经传递有赖于突触蛋白的完整性[20],突触蛋白Ⅰ被降解破坏后突触传递功能发生异常。本研究中发现钙蛋白酶可降解突触蛋白Ⅰ,提示钙蛋白酶对突触蛋白Ⅰ的降解可能损伤神经元正常突触功能,破坏神经递质的正常释放和再摄取,加重脑出血后神经功能缺失。PC12细胞是大鼠肾上腺嗜铬细胞瘤细胞,经神经生长因子处理后表现出神经元的特性,如停止分裂增殖、生长出突起等[21]。在体外培养的神经元样PC12细胞中,外源性钙蛋白酶导致细胞活力下降,钙蛋白酶抑制能够延缓钙蛋白酶的损伤作用,提示钙蛋白酶对神经元存在直接损伤作用,既往研究发现在乳胞素介导的原代皮层神经元损伤中,钙蛋白酶被激活,降解血影蛋白,参与神经损伤,而钙蛋白酶抑制剂具有神经保护作用,该结果与本研究的结果相契合[22]。
本研究的不足之处在于并未深入探讨钙蛋白酶的其他底物在脑出血后神经元损伤中的作用,因此有必要在突触蛋白Ⅰ基因敲除小鼠中进一步探讨钙蛋白酶对神经元损伤作用的具体机制。因此,钙蛋白酶通过降解突触蛋白Ⅰ导致神经元损伤,参与脑出血后神经功能缺失。因此,钙蛋白酶有望成为脑出血后针对神经损伤的治疗靶点。
1 Gao C, Du H, Hua Y, et al. Role of red blood cell lysis and iron in hydrocephalus after intraventricular hemorrhage[J]. J Cereb Blood Flow Metab, 2014, 34:1070-1075.
2 Lauer A, Pfeilschifter W, Schaffer CB, et al. Intracerebral haemorrhage associated with antithrombotic treatment:translational insights from experimental studies[J].Lancet Neurol, 2013, 12:394-405.
3 Bernath E, Kupina N, Liu MC, et al. Elevation of cytoskeletal protein breakdown in aged Wistar rat brain[J]. Neurobiol Aging, 2006,27:624-632.
4 Kataya ma J, Akaike N, Nabekura J.Characterization of pre- and post-synaptic metabotropic glutamate receptor-mediated inhibitory responses in substantia nigra dopamine neurons[J]. Neurosci Res, 2003,45:101-115.
5 Lee MS, Kwon YT, Li M, et al. Neurotoxicity induces cleavage of p35 to p25 by calpain[J].Nature, 2000, 405:360-364.
6 Krafft PR, Rolland WB, Duris K, et al. Modeling intracerebral hemorrhage in mice:injection of autologous blood or bacterial collagenase[J]. J Vis Exp,2012:e4289.
7 Xi G, Keep RF, Hoff JT. Mechanisms of brain injury after intracerebral haemorrhage[J].Lancet Neurol, 2006, 5:53-63.
8 Belur PK, Chang JJ, He S, et al. Emerging experimental therapies for intracerebral hemorrhage: targeting mechanisms of secondary brain injury[J]. Neurosurg Focus,2013, 34:E9.
9 Zheng M, Gong Y, Wang X, et al. Alteration of intracellular calcium and its modulator SLC24A6 after experimental intracerebral hemorrhage[J]. Acta Neurochir Suppl, 2013,118:169-173.
10 Rami A, Krieglstein J. Protective effects of calpain inhibitors against neuronal damage caused by cytotoxic hypoxia in vitro and ischemia in vivo[J]. Brain Res, 1993, 609:67-70.
11 Ono Y, Sorimachi H. Calpains:An elaborate proteolytic system[J]. Biochim Biophys Acta,2012, 1824:224-236.
12 Storr SJ, Carragher NO, Frame MC, et al. The calpain system and cancer[J]. Nat Rev Cancer,2011, 11:364-374.
13 Amini M, Ma CL, Farazifard R, et al.Conditional disruption of calpain in the CNS alters dendrite morphology, impairs LTP,and promotes neuronal survival following injury[J]. J Neurosci, 2013, 33:5773-5784.
14 Moldoveanu T, Hosfield CM, Lim D, et al. A Ca(2+) switch aligns the active site of calpain[J]. Cell, 2002, 108:649-660.
15 Cesca F, Baldelli P, Valtorta F, et al. The synapsins:key actors of synapse function and plasticity[J]. Prog Neurobiol, 2010, 91:313-348.
16 Rosahl TW, Spillane D, Missler M, et al.Essential functions of synapsins I and II in synaptic vesicle regulation[J]. Nature, 1995,375:488-943.
17 Murrey HE, Gama CI, Kalovidouris SA, et al.Protein fucosylation regulates synapsin Ia/Ib expression and neuronal morphology in primary hippocampal neurons[J]. Proc Natl Acad Sci U S A, 2006, 103:21-26.
18 Gitler D, Xu Y, Kao HT, et al. Molecular determinants of synapsin targeting to presynaptic terminals[J]. J Neurosci, 2004,24:3711-3720.
19 Orlando M, Lignani G, Maragliano L, et al.Functional role of ATP binding to synapsin I in synaptic vesicle trafficking and release dynamics[J]. J Neurosci, 2014, 34:14752-14768.
20 Chi P, Greengard P, Ryan TA. Synaptic vesicle mobilization is regulated by distinct synapsin I phosphorylation pathways at different frequencies[J]. Neuron, 2003, 38:69-78.
21 Chen TI, Chiu HW, Pan YC, et al. Intermittent
【点睛】
本文通过在体和离体模型的研究观察到钙蛋白酶通过降解其底物突触蛋白Ⅰ参与神经损伤的作用及其机制,为脑出血后继发性神经损伤寻找新的治疗靶点提供依据。
