双(4-氟苯基)-3′-二乙氧基膦酰苯基氧膦的合成与表征
2014-02-18张成凤汤红英张中标
张成凤,汤红英,张中标
(天津师范大学 天津市水资源与水环境重点实验室,天津 300387)
含膦酰基团的高分子化合物耐高温,抗氧化性能好,在高温和干、湿状态下均有良好的质子传导性和低吸水性,能够有效减少膜的溶胀,可应用于燃料电池高温操作时质子交换膜的制备.同时,此类化合物具有较高的热稳定性和化学稳定性,尤其是在较宽的pH范围内具有超强的抗氯性能,因此也有望成为新型的反渗透纳滤水处理膜材料.由于合成具有高传导性能的高膦酰化聚合物难度大,因此对于此类聚合物材料的研究相对较少[1-4].目前合成方法有2种:一种是对溴化或锂化的聚合物进行改性,这种方法取得了较好结果,但是膦酰化程度不易控制,膦酰化过程中高分子链易断裂;另一种方法是将含有膦酸基团的单体与其他适合的单体进行直接聚合,而膦酸基团一般位于脂肪链、二酚等电子云密度大的单体上,因此聚合效果较差,大部分是低聚物[5-7].
本课题组设计合成了一种新型的膦酰化单体:双(4-氟苯基)-3′-二乙氧基膦酰苯基氧膦.该单体与不含膦酸基团的活化的芳香二卤化物和二酚,在高沸点非质子有机溶剂中,在中强碱的存在下,与其他单体发生聚合反应,得到含有膦酸酯基团的新型聚合物.这一聚合物经过酸化及中和处理,分别得到含有膦酸基团和膦酸盐基团的新型高分子材料,从而为此种材料的反渗透除盐和质子传导性能等方面的研究提供基础.
1 实验
1.1 仪器与试剂
仪器:Varian 400型核磁共振仪(CDCl3、D2O或DMSO-d6为溶剂,TMS为内标),由美国Varian公司生产;WRR型数字显示显微熔点测定仪,由上海物光科技开发有限公司生产;IR-435型红外光谱仪(KBr压片法),由日本岛津公司生产;CHN corder MT-3型元素分析仪,由日本Yanaco公司生产.
试剂:实验所用试剂均为AR级.双(4-氟苯基)苯基氧膦(BFPPO),由天津砚津科技有限公司生产;无水乙醇、浓硫酸、浓硝酸、氯化亚锡、亚硝酸钠、碘化钾,均由天津北方天医化学试剂厂生产;三乙胺、亚磷酸二乙酯、甲苯,由天津金玻化试验设备有限公司提供,亚磷酸二乙酯重新蒸馏后使用,甲苯及三乙胺经过无水处理;Pd(PPh3)4由课题组自行合成.
1.2 膦酰化单体的制备
膦酰化单体的合成路线如下:
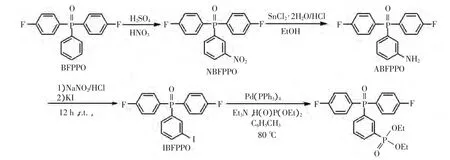
1.2.1 双(4-氟苯基)-3′-硝基苯基氧膦(NBFPPO)的合成[8]
室温和氮气氛围下,将22.79g的BFPPO和0.2L浓硫酸混合,搅拌至均相,冰浴(0~5℃).将7.51g的浓硝酸和28.0mL的浓硫酸混合液滴加至反应体系中,滴加完毕后自然升温至室温,反应2h(TLC监测反应).反应完成后将反应液倒入3 L冰水中,过滤,滤饼用三氯甲烷溶解.然后依次分别用水、饱和碳酸钾溶液、饱和氯化钠溶液各洗2次,无水硫酸镁干燥过夜,真空去溶剂.最终得到黄色固体23.96g,m.p.151~151.5℃(lit.151~151.5℃),产率93%(lit.92%).
1.2.2 双(4-氟苯基)-3′-氨基苯基氧膦(ABFPPO)的合成[8]
室温和氮气氛围下,将21.42g的NBFPPO和306.0mL的无水乙醇混合,搅拌加热回流使反应液呈均相.在分批多次加入氯化亚锡54.00g的同时,分3次加入浓盐酸(每次33.0mL),加完后继续反应2.5h.自然冷却至室温,将反应液倒入3 L冰水中.用质量分数40%的氢氧化钠溶液将体系调至pH为13,过滤.滤饼用三氯甲烷溶解后,依次用饱和碳酸钠溶液、饱和氯化钠溶液洗涤,无水硫酸镁干燥过夜.真空去溶剂后得到微黄色固体15.71g,m.p.148.0~150.0℃(lit.148.5~150.3℃),产率82%(lit.80%).
1.2.3 双(4-氟苯基)-3′-碘苯基氧膦(IBFPPO)的合成[8-9]
室温下将3.29g的ABFPPO、浓盐酸/水混合液(18.5mL浓盐酸、2.0mL水)混合,搅拌至均相.冰浴下滴加1.14g亚硝酸钠的水(7.5mL)溶液.滴加完毕后,冰浴继续反应0.5h,反应液趁冷过滤后滴加至碘化钾溶液中(7.28g KI,18.5mL水).然后自然升温至室温,搅拌反应过夜,反应液用90.0mL乙酸乙酯萃取3次,合并有机相.有机相用质量分数为10%的氢氧化钠溶液洗涤2次,再用饱和氯化纳溶液洗涤,无水硫酸镁干燥过夜,真空去溶剂得粗品,色谱柱层析得到淡黄色固体 3.58g,m.p.108.6~110.0℃(lit.108.4~110.0℃),产率85%(lit.85%).
1.2.4 双(4-氟苯基)-3′-二乙氧基膦酰苯基氧膦的合成
在氮气氛围下,将4.20g的IBFPPO和1.15g的Pd(PPh3)4加入到三口圆底烧瓶中,置换气3次后,加入溶剂甲苯12.0mL、三乙胺5.0mL、亚磷酸二乙酯2.6mL,加热升温至80℃,反应过夜(TLC监测反应).当原料IBFPPO反应完,停止反应,过滤,滤液真空去溶剂,色谱柱层析(石油醚和乙酸乙酯的体积比为1∶2),得到浅黄色油状物3.24g,收率72%.
在合成双(4-氟苯基)-3′-二乙氧基膦酰苯基氧膦过程中,加入固体原料后一定要用惰性气体置换气3次,并且保证反应是在惰性气体氛围下进行,无水、无氧.催化剂Pd(PPh3)4为新制的亮黄色固体,放置时间过长会发生氧化,影响催化效果.
双(4-氟苯基)-3′-二乙氧基膦酰苯基氧膦的结构表征:1HNMR(CDCl3,400MHz)δ:1.28(t,CH3,J=6.0Hz),4.08(m,CH2),7.15~7.20(dt,4Harom,J=2.0Hz,6.4,8.4),7.61~7.68(m,5Harom),7.83~7.88(m,1Harom),7.98~8.06(m,2Harom);31P NMR(CDCl3,400MHz)δ:16.42,26.84;13C NMR(CDCl3,400MHz)δ :16.2,62.4,115.9,116.4,127.0,128.4,128.5,128.6,129.0,134.3,135.7,163.6,166.9;H-MS:[M+Na]+473.0853,cal.473.0854.
2 结果与分析
以氘代氯仿作溶剂,利用Varian 400型核磁共振仪进行扫描,各化合物的NMR信号得到了合理的归宿;H-RMS以傅里叶变换离子回旋共振质谱仪(FTICR-MS)测定,H-MS:[M+Na]+473.0853,cal.473.0854,显示该单体结构完全正确.
1HNMR图谱如图1所示.
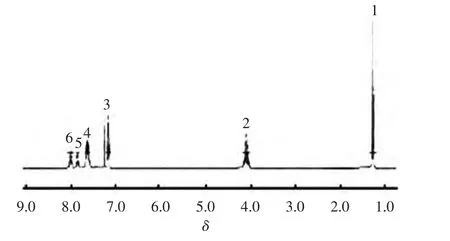
图1 膦酰化单体的1H NMR图谱Fig.1 1H NMR spectrum of phosphonylation monomer
由图1可知,化学位移1.26~1.29为1位置甲基,三重峰,6H,耦合常数J=6,δ4.05~4.16为与甲基相连的2位置亚甲基多重峰,4H,δ7.15~7.20为芳环上3位置的CH,双三重峰,4H,耦合常数J=2.0、6.4、8.4,δ7.83~7.88为芳环上4位置的CH,多重峰,5H,化学位移7.98~8.00为芳环上5位置CH多重峰,1H,δ8.01~8.06为芳环上 6位置CH,多重峰,2H,δ7.26为氯仿溶剂峰.核磁共振1H NMR表征的结果与产品的理论结构一致.
31P NMR图谱如图2所示.化学位移26.84为a位置P=O上P的峰,16.42为b位置膦酸酯基上P的峰,核磁共振31P NMR表征的结果与产品的理论结构一致.
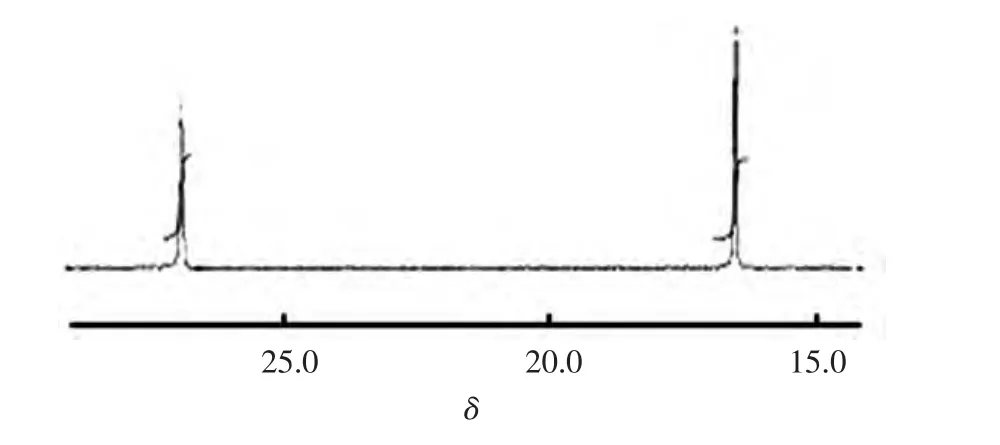
图2 膦酰化单体的31P NMR图谱Fig.2 31P NMR spectrum of phosphonylation monomer
13C NMR图谱如图3所示.化学位移16.2为1位置膦酸酯基团上甲基2C峰,62.4为2位置膦酸酯基团上亚甲基2C峰,115.9、116.1、116.2、116.3为芳环3位置的4C峰,126.9为芳环4位置上1C峰,128.4、128.6、128.9为芳环上 5位置 3C峰,134.3、134.5、134.6、134.7、134.9、135.2、135.3、135.4、135.5、135.6、135.7为芳环上 6、7、8位置 8C 峰,163.6、166.9为芳环上9位置2C峰,77.0、77.2、77.4为氘代氯仿的溶剂峰.核磁共振13C NMR表征的结果与产品的理论结构一致.

图3 膦酰化单体的13C NMR图谱Fig.3 13C NMR spectrum of phosphonylation monomer
3 结论
本研究以双(4-氟苯基)苯基氧膦(BFPPO)为原料,通过硝化、还原胺化、碘代、钯催化-偶联,得到新型的膦酰化单体双(4-氟苯基)-3′-二乙氧基膦酰苯基氧膦,纯度超过99.5%.采用核磁共振、熔点仪、HRMS(ESI)等实验方法对产物结构进行表征,结果表明本研究合成化合物的结构与理论结构一致.
[1]MIYATAKE K J,HAY A S.New poly(arylene ether)s with pendant phosphonic acid groups[J].J Polym Sci Part A:Polym Chem,2001,39(21):3770-3779.
[2] JAKOBY K,PEINEMANN K V,NUNES S P.Palladium-catalyzed phosphonation of polyphenylsulfone[J].Macromol Chem Phys,2003,204(1):61-67.
[3] LIU B J,ROBERTSON G P,GUIVER M D,et al.Fluorinated poly(arylether)containing a 4-bromophenyl pendant group and its phosphonatedderivative[J].MacromolRapidCommun,2006,27(17):1411-1417.
[4] LAFITIE B,JANNASCH P.Polysulfone ionomers functionalized with benzoyl(difluoromethylenephosphonic acid)side chains for proton-conducting fuel-cell membranes[J].J Polym Sci Part A:Polym Chem,2007,45(2):269-283.
[5] BOUTEVIN B,HAMOUI B,PARISIJ P,et al.Homopolymerization and copolymerization of salt formed from a new diethyl styrenic phosphonate monomer[J].Eur Polym J,1996,32(2):159-163.
[7]TROTTA F,CERESA M S,MARTINA K,et al.New poly ether ether ketones containing phosphorus for membrane preparation[J].Asia-Pac J Chem Eng,2010,5(1):249-255.
[8]PAPADIMITRIOU K D,AIKATERINI A K,KALLITSIS J K.Phosphonated fully aromatic polyethers for PEMFCs applications[J].J Polym Sci Part A:Polym Chem,2010,48(13):2817-2828.
[9] 刘小斌,汤红英,张中标.新型碘代氧膦单体的合成[J].精细化工中间体,2011,4(2):48-50.
[10]SOHN J,KIBURZ B,LI Z T,et al.Inhibition of Cdc25phosphatases by indolyldihydroxyquinones[J].J Med Chem,2003,46(13):2580-2588.
