分散固相萃取净化-液相色谱-质谱联用测定水产苗种中硝基呋喃类代谢物残留量
2014-01-17严忠雍张小军梅光明李佩佩
严忠雍,张小军,*,梅光明,李佩佩,龙 举,祝 银,王 建
分散固相萃取净化-液相色谱-质谱联用测定水产苗种中硝基呋喃类代谢物残留量
严忠雍1,2,张小军1,2,*,梅光明1,2,李佩佩1,2,龙 举1,2,祝 银1,2,王 建3
(1.浙江省海洋水产研究所,浙江 舟山 316021;2.浙江省海洋渔业资源可持续利用技术研究重点实验室,浙江 舟山 316021;3.浙江工商大学食品与生物工程学院,浙江 杭州 310035)
建立分散固相萃取净化-液相色谱-质谱法测定水产苗种中氨基脲、5-甲基吗啉-3-氨基-2-噁唑烷基酮、3-氨基-2-噁唑烷基酮、1-氨基-2-内酰脲,4 种硝基呋喃类代谢物残留量的方法。样品经甲醇-水(8∶1,V/V)溶液洗涤后,盐酸溶液水解,以2-硝基苯甲醛作为衍生化试剂,乙酸乙酯萃取,浓缩,分散固相萃取净化,采用液相色谱-质谱仪、多反应监测扫描模式检测和同位素内标定量。4 种硝基呋喃类代谢物在0.5~20 μg/L范围内线性相关系数大于0.997,检出限0.1 μg/kg,回收率90.5%~104.5%,相对标准偏差1.56%~8.08%。本方法灵敏、高效、简单、重复性好,满足硝基呋喃类代谢物的检测要求。
分散固相萃取;液相色谱-质谱;水产苗种;硝基呋喃类代谢物
硝基呋喃类药物是一类合成的抗菌药物,主要包括呋喃妥因、呋喃它酮、呋喃唑酮、呋喃西林,作用于微生物酶系统,抑制乙酰辅酶A,干扰微生物糖类的代谢,从而起抑菌作用,广泛用于家禽、家畜、水产等动物传染病的预防与治疗[1]。由于硝基呋喃类药物对人类健康具有潜在危害,是世界各国普遍禁止使用的药物[2]。我国于2002年颁布了禁止使用硝基呋喃类抗生素的禁令[3]。硝基呋喃类抗菌剂原型药在动物体内代谢迅速,但是其代谢产物和蛋白质结合后很稳定[4]。目前,欧盟国家要求代谢产物为目标分析物,以达到检测硝基呋喃残留的目的[5]。硝基呋喃类代谢物有1-氨基-2-内酰脲(1-aminohydantoin,AHD)、5-甲基吗啉-3-氨基-2-噁唑烷基酮(5-morpholinomethyl-3-amino-2-oxazolidone,AMOZ)、3-氨基-2-噁唑烷基酮(3-amino-2-oxazolidone,AOZ)、氨基脲(semicarbazide,SEM)。
目前文献报道的硝基呋喃类代谢物残留的分析方法主要是:高效液相色谱法[6]和液相色谱-质谱法[7-11];硝基呋喃类代谢物检测的前处理方法有固相萃取[12-14]及液液萃取[15]。原有的文献方法能基本满足水产品中硝基呋喃类代谢物的检测分析要求,但在检测水产苗种时并不能达到较好的效果。水产苗种异于水产成品,尤其是虾蟹类苗种,在其苗种期个体幼小通常带壳制样,水解过程中消耗大量盐酸,导致水解不完全,衍生化效率降低;水产苗种样品通常含水率较多,且样品基质成分复杂,如果未进行合适有效净化手段,会造成分析物严重损失,因此有必要建立一种针对水产苗种中硝基呋喃类代谢物的分析方法。分散固相萃取法是一种快速、稳定的前处理方法,但用于硝基呋喃类代谢物检测尚未见报道。本研究在前处理阶段根据盐酸消耗量优化盐酸用量,利用分散固相萃取净化结合液相色谱-质谱法测定,建立水产苗种中硝基呋喃类代谢物残留量分析方法,本方法简便、高效、快速、灵敏度高、重复性好,定量限、回收率与精密度均满足残留分析和残留限量要求,同时对其他产品中硝基呋喃类代谢物的检测具有参考意义。
1 材料与方法
1.1 材料与试剂
盐酸、磷酸氢二钾(均为分析纯) 国药集团化学试剂有限公司;乙酸乙酯、乙酸铵、甲酸、二甲亚砜、甲醇、2-硝基苯甲醛等(均为色谱纯),硝基呋喃代谢物标准品及其内标物:AOZ、SEM-HCl、AHD-HCl、AMOZ、AOZ-D4、SCA-HCl-13C-15N2、AHD-HCl-13C3、AMOZ-D5(纯度均≥98%) 美国Sigma公司;N-丙基乙二胺(N-propylethylendiamine,PSA)、十八烷基键合硅胶吸附剂(C18)、弗罗里硅土(Florisil)、中性氧化铝(Al2O3)、多壁碳纳米管(MWCNT) 天津Agela有限公司。
标准储备液:准确称取5.00 mg AOZ、SEM、AHD、AMOZ标准品(SEM、AHD的质量均为SEM-HCl,AHDHCl去除盐酸后换算的质量),用甲醇溶解定容于50 mL容量瓶,4 ℃避光保存,保存期限为6 个月;使用时逐级稀释至100 ng/mL。
1.2 仪器与设备
ACQUITy超高效液相色谱-质谱仪(配电喷雾离子源,Quattro Premier XE) 美国Waters公司;T18匀浆机、MS2漩涡混合器 德国IKA公司;氮气吹干仪 美国Organomatio公司;Centrifuge5810高速离心机 德国Eppendorf公司;SK25IOHP超声波清洗器 上海科导超声仪器有限公司;恒温振荡器 常州国华电器有限公司。
1.3 方法
1.3.1 样品处理
准确称取2.00 g均质水产苗种样品于50 mL离心管中,加1 mL水和8 mL甲醇,涡旋振荡2 min,6 000 r/min离心5 min,弃去上清液。加100 ng/mL混合内标工作液0.1 mL,再加0.2 mol/L盐酸溶液5 mL(当样品是虾蟹类苗种时,改加0.9 mol/L盐酸溶液5 mL)和0.05 mol/L 2-硝基苯甲醛0.15 mL,涡旋振荡2 min后,37 ℃恒温避光振荡16 h。取出离心管冷却到室温,加入适量1 mol/L磷酸氢二钾溶液,调节pH值至7.0~7.5。加入5 mL乙酸乙酯,涡旋振荡1 min,6 000 r/min离心5 min,转移上清液至15 mL聚四氟乙烯离心管中;下层溶液加入5 mL乙酸乙酯重复提取一次,合并后的上清液40 ℃ N2吹干。残渣用1 mL流动相溶解,加100 mg PSA,涡旋1 min,6 000 r/min离心3 min,取上清液经0.22 μm有机滤膜过滤,转移至进样瓶供液相色谱-质谱分析。
1.3.2 色谱条件
色谱柱:ACQUITy UPLC BEH C18柱(2.1 mm× 50 mm,1.7 μm);柱温40 ℃;样品室温度10 ℃;进样量10 μL;流速0.2 mL/min;流动相A为含有体积分数0.1%甲酸的2 mmol/L醋酸铵溶液,B为甲醇,梯度洗脱(线性改变),梯度洗脱设置详见表1。

表1 流动相梯度洗脱Table 1 Mobile phase composition for gradient elution
1.3.3 质谱条件
离子源:电喷雾离子(electorspray ionization,ESI+)源,正离子扫描;检测方式:多重反应监测(multiple reaction monitoring,MRM);毛细管电压3.0 kV;离子源温度120 ℃;脱溶剂气温度380 ℃;脱溶剂气流量600 L/h;锥孔气流量50 L/h;锥孔电压和碰撞能量等质谱多反应监测实验条件见表2。
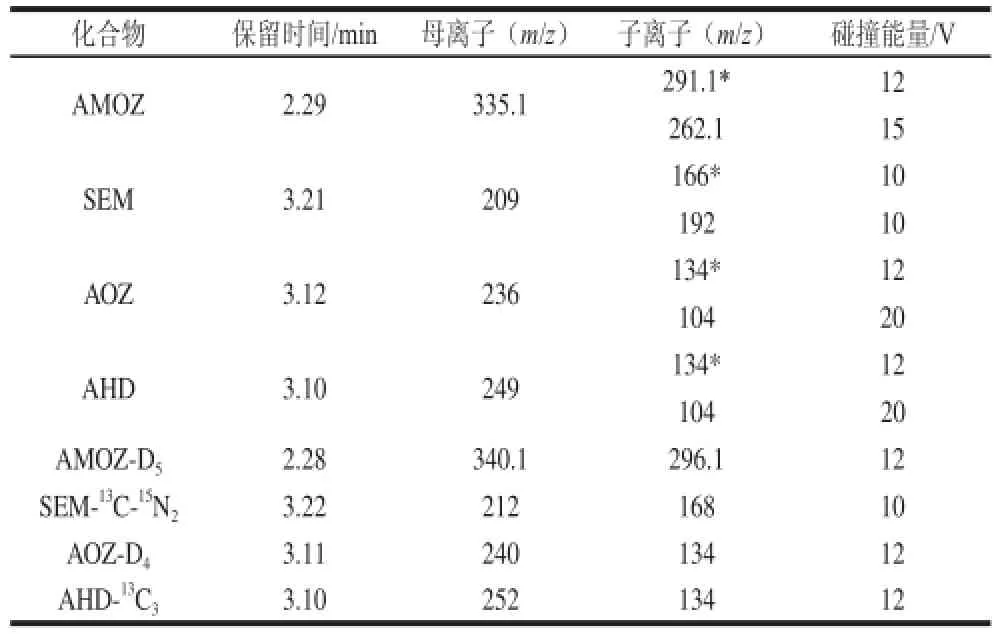
表2 硝基呋喃代谢物和相应内标的质谱参数Table 2 ESI-MS-MS conditions for the analysis of 4 nitrofuran metabolites and their corresponding internal standards
2 结果与分析
2.1 样品洗涤
水产苗种样品含水率较多,且样品基质成分复杂,若当水产成品按农业部783号公告-1—2006《水产品中硝基呋喃类代谢物残留量的测定:液相色谱-串联质谱法》[16]前处理,直接影响检测结果。若对水产苗种先用甲醇-水混合溶液(8∶1,V/V)洗涤处理[17],可以去除样品均质液中黏液、色素等杂质,减少分析干扰,防止被测物和内标物丢失。加标水平为2.5 μg/kg梭子蟹苗种未洗涤处理的色谱图及洗涤处理后的色谱图如图1、2所示。对比可知,未进行洗涤处理的样品AHD未出峰,AHD-13C3、AOZ-D4,AOZ峰面积小,响应值低;而SEM-13C-15N2未分开,峰形杂乱。而经洗涤处理的样品基线较平稳,除AHD峰面积较小外,其他目标物响应值高,峰形完整。
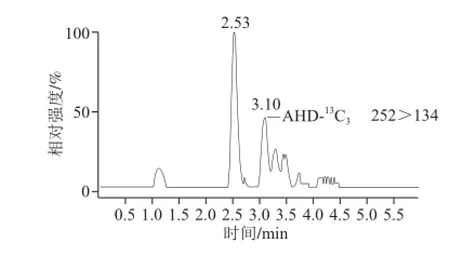

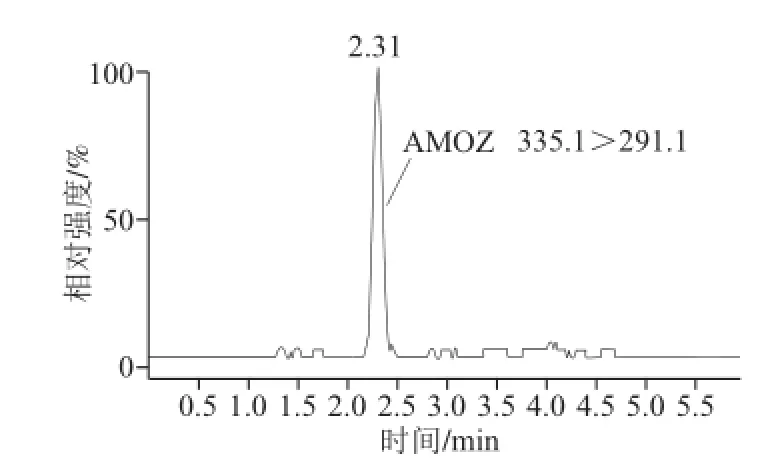
图1 未洗涤处理的2.5μg/kg梭子蟹苗种MRM图谱Fig.1 MRM ion chromatograms of crab fingerlings (2.5 μg/kg) without washing

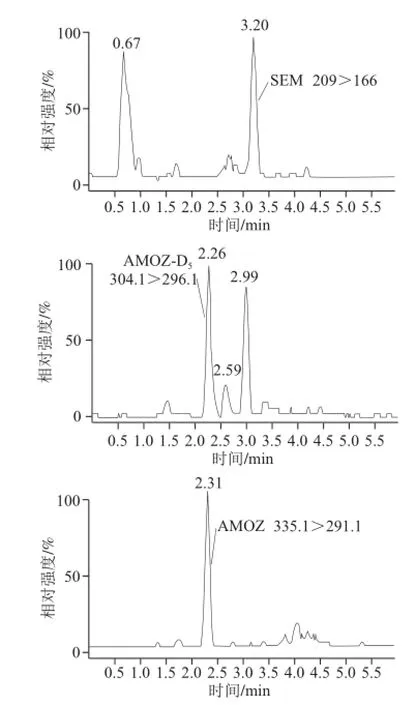
图2 洗涤处理后的2.5μg/kg梭子蟹苗种MRM图谱Fig.2 MRM ion chromatograms of crab fingerlings (2.5 μg/kg) with washing
2.2 盐酸用量优化
虾蟹类苗种由于个体较小,不易去壳,通常带壳制样,而壳的碳酸钙含量很高,能与盐酸反应,使反应体系的酸度发生变化,导致酸水解不完全,衍生化效率降低。实验分别取5 mL 0.2、0.4、0.6、0.8 mol/L和1.0 mol/L盐酸溶液水解加标水平为2.5 μg/kg的梭子蟹苗种与梭子蟹肌肉样品,测定酸水解16 h后的pH值,比较不同浓度盐酸水解下的回收率,衡量碳酸钙与盐酸反应的消耗量,以得到虾蟹类苗种水解所需盐酸的最佳值。结果表明,随着 盐酸溶液浓度的增大对梭子蟹肌肉中的4 种硝基呋喃代谢物并不造成影响,5 mL 0.2 mol/L盐酸溶液足以完全水解肌肉中的蛋白质,且有较好的衍生化效果,无需过量提高盐酸浓度。不同浓度的盐酸溶液对梭子蟹苗种中AMOZ和AOZ影响不大,无明显变化趋势;随着盐酸浓度增大,梭子蟹苗种中AHD峰面积逐渐增大后趋于平缓,表明之前蛋白质并未充分水解;而梭子蟹苗种中SEM浓度的变化略带波动性,因SEM是甲壳类水产品中的内源性物质,自然存在于甲壳[18]。表3为酸水解16 h后测定的pH值,由于在梭子蟹肉中5 mL 0.2 mol/L盐酸溶液足以完全水解肌肉中的蛋白质,表明酸水解后pH 2.2是样品充分酸水解的标示;而当样品是梭子蟹苗种时,酸水解后pH 2.2对应的盐酸浓度约为0.9 mol/L。最终确定5 mL 0.9 mol/L盐酸作为虾蟹类苗种酸水解所需的盐酸量,其水解后测定色谱图见图3。
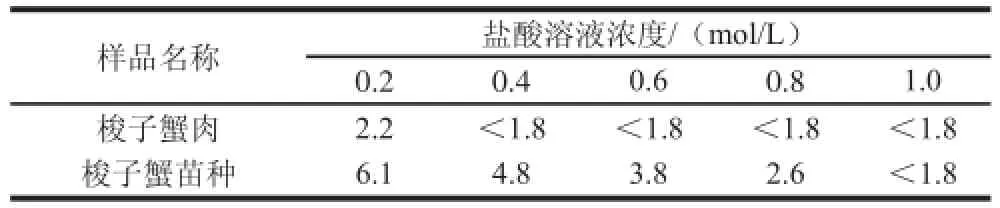
表3 样品在不同浓度盐酸水解16 h后测定的pH值Table 3 pH values of samples after 16 h of hydrolysis with different concentrations of hydrochloric acid
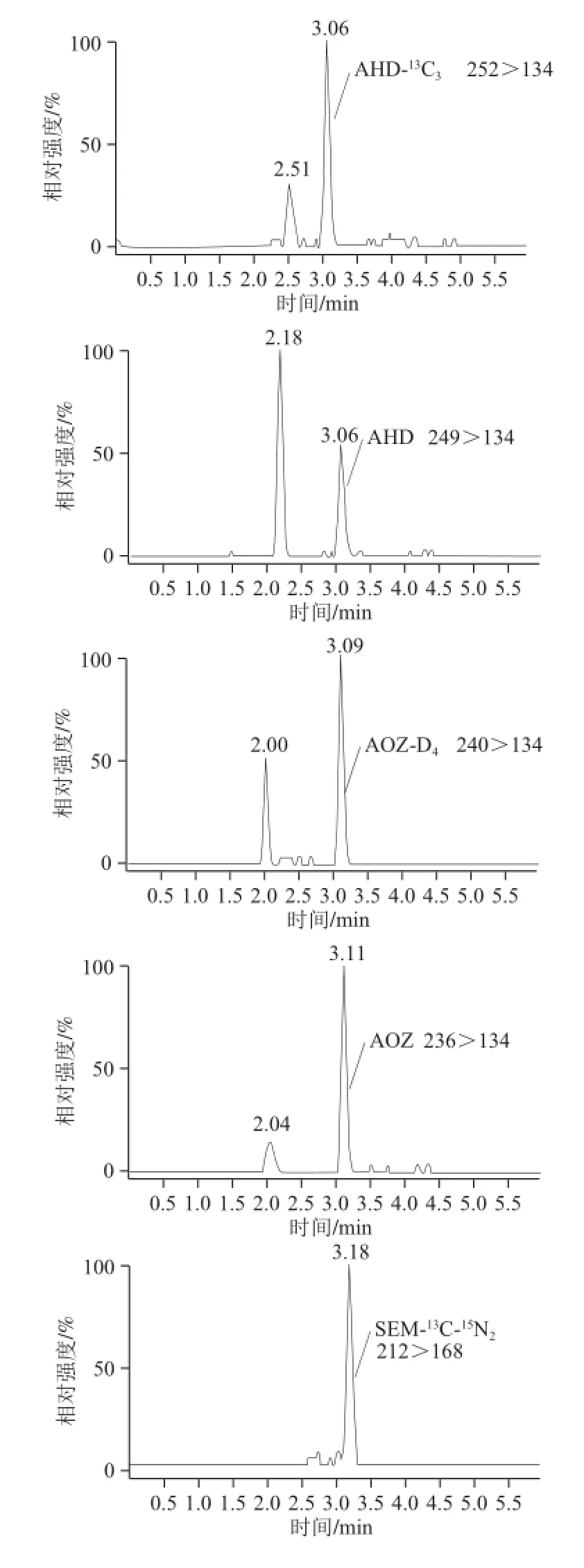

图3 100 mg PSA净化后2.5μg/kg的梭子蟹苗种MRM图Fig.3 MRM ion chromatograms of 2.5 μg/kg crab fingerlings purified with 100 mg of PSA
2.3 吸附剂的选择和优化
水产苗种样品脂类、色素等杂质较多,浓缩溶解后,溶液呈混浊状,需净化处理后上样。实验分别称取一 定量的PSA、C18、Florisil、Al2O3、MWCNT净化加标水平为2.5 μg/kg的梭子蟹苗种样品,考察不同吸附剂的净化效果,比较各药物的回收率,结果如图4所示。MWCNT具有巨大的比表面积和结构特异性,能有效去除色素类等干扰物[19],但对AMOZ、SEM、AHD及AOZ的吸附作用也很强;Florisil作为强极性吸附剂,无法有效去除脂类杂质,且对AMOZ具较强的吸附性。而PSA、C18、Al2O3对AMOZ、SEM、AHD和AOZ都有较高的回收率,为得到更好的净化效果和回收率,对这3 种吸附剂用量进行优化。分别称取25、50、75、100 mg和125 mg的PSA、C18、Al2O3净化加标水平为2.5 μg/kg的梭子蟹苗种样品,观察净化液的颜色,透明度,并计算信噪比(以AMOZ为例),如图5所示。Al2O3和C18虽有较好的回收率,能去除脂类杂质[20],但净化效果不明显,即使加大吸附剂用量,也未得到良好的改善,净化液仍是混浊状。PSA作为弱阴离子交换剂,能去除脂肪酸、甾醇类、有机碳水化合物、色素等杂质[21],净化效果好,净化液呈无色透明状,且有较高的回收率。当PSA用量小于100 mg时,净化效果随用量增大而明显增大;而超过100 mg后,趋于平缓。最终选择100 mg的PSA作为吸附剂,降低样品基质中色素、碳水化合物、脂类等物质,也获得净化效果和回收率之间较好的平衡。
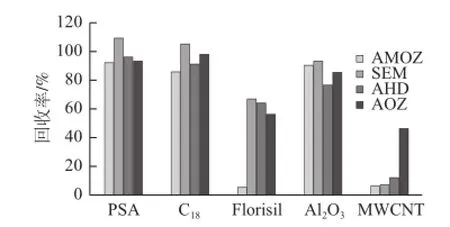
图4 吸附剂对2.5μg/kg梭子蟹苗种中4种硝基呋喃代谢物回收率的影响Fig.4 Influence of different sorbents on the recoveries of 4 nitrofuran metabolites in 2.5 μg/kg crab fingerlings
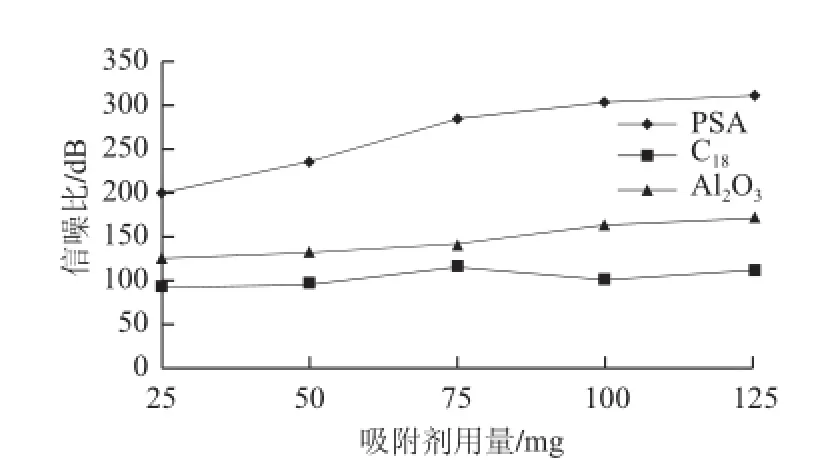
图5 吸附剂用量对2.5μg/kg梭子蟹苗种中AMOZ响应值的影响Fig.5 Influence of different sorbent dosages on the SNR of AMOZ in 2.5 μg/kg crab fingerlings
2.4 线性范围及检出限
用硝基呋喃4 种代谢物标准使用液配制质量浓度分别为0.5、1.0、2.0、5.0、10.0、20.0 μg/L的标准溶液,内标添加质量浓度均为5.00 μg/kg,进样后制作标准曲线,硝基呋喃4 种代谢物的线性方程及相关系数如表4所示。结果表明,硝基呋喃4 种代谢物在0.5~20.0 μg/L内线性良好,方法的检出限理论上为被测物质丰度较弱子离子的信噪比(RSN)不小于3,将加标质量浓度逐级稀释添加于空白样品中测定信噪比,最终确定该方法的4 种硝基呋喃类代谢物检出限均为0.1 μg/kg,定量限均为0.3 μg/kg。

表4 4 种硝基呋喃代谢物的线性方程与相关系数Table 4 Linear equations and correlation coefficients of 4 nitrofuran metabolliitteess
2.5 回收率及精密度
准确称取2.00 g阴性乌鳢苗种样品,添加水平为0.5、1.0、5.0 μg/kg的标准溶液,每个添加水平平行测定6 次,计算其回收率和精密度,见表5。结果表明,方法在0.5~5.0 μg/kg添加水平之间的回收率为90.5%~104.5%,相对标准偏差为1.56%~8.08%。完全符合硝基呋喃类代谢物的检测要求。2.6 实际样品测定
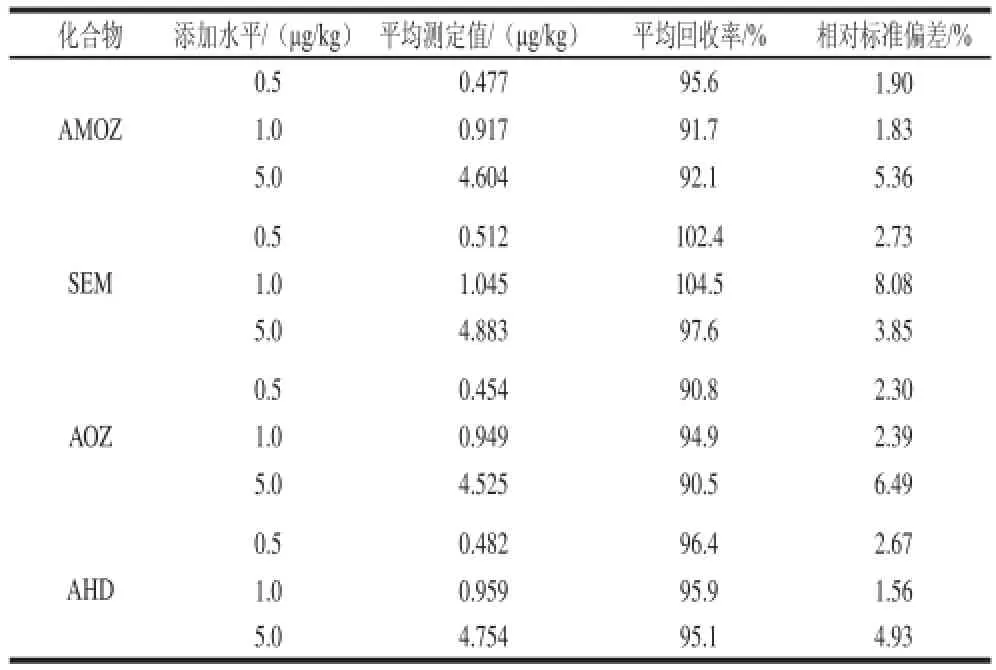
表5 4 种硝基呋喃代谢物的平均回收率和相对标准偏差(n=6)Table 5 Average recoveries and relative standard deviations for 4 nitrofuran metabolitess (n=6)
在实际测定的34 个苗种样品(采自不同养殖场)中包括12 个乌鳢苗种、12 个南美白对虾苗种和10 个梭子蟹苗种,检出8 个阳性样品。其中阳性乌鳢苗种1 个,SEM含量1.12 μg/kg;阳性对虾苗种1 个,AOZ含量2.05 μg/kg;阳性梭子蟹苗种6 个,SEM含量1.24、1.32、1.12、1.40、1.06μg/kg和1.17 μg/kg。由于SEM是甲壳类水产品中的内源性物质,呋喃西林并不是甲壳类水产品中SEM的唯一来源,故6 个阳性梭子蟹苗种不能直接作为是否进行投药依据;而阳性乌鳢苗种和阳性对虾苗种表明部分水产苗种在生长期就已开始投放硝基呋喃类药物。因此,有必要在水产苗种阶段控制硝基呋喃类药物,为水产品质量安全提供重要保障。
3 结 论
有关硝基呋喃类代谢物残留量测定的文献报道很多,但大多数研究对象为水产成品,针对水产苗种的研究较少,而原有的文献方法用于水产苗种检测并未有较好的效果。为满足水产苗种中硝基呋喃类代谢物检测的需要,根据水产苗种的差异性和特殊性,建立了分散固相萃取净化-液相色谱-质谱法测定水产苗种中硝基呋喃类代谢物的分析方法,前处理优化盐酸用量,采用PSA作为吸附剂净化分析物,方法重复性好,灵敏度高,满足硝基呋喃类代谢物的检测要求。
参考文献:
[1] 葛宝坤, 王云凤, 常春艳, 等. 测定鸡肉、水产品中四种硝基呋喃类药物残留量的固相萃取-液相色谱法[J]. 分析测试学报, 2003, 22(5): 91-93.
[2] 孙涛, 胡开峰, 乔昆云, 等. 超高效液相色谱-串联质谱法测定水产品中硝基呋喃代谢物残留[J]. 分析仪器, 2010, 29(5): 27-31.
[3] 彭涛, 储晓刚, 杨强, 等. 高效液相色谱/串联质谱法测定奶粉中硝基呋喃代谢物[J]. 分析化学, 2005, 33(8): 1073- 1076.
[4] 张秀妍, 马琳, 王慧龙. 海参和海参苗种中硝基呋喃类代谢物残留的液质联用检测法[J]. 口岸卫生控制, 2012, 17(6): 24-28.
[5] 林黎明, 林回春, 刘心同, 等. 固相萃取高效液相色谱-质谱法测定动物组织中硝基呋喃代谢产物[J]. 分析化学, 2005, 33(5): 707-710.
[6] 王媛, 蔡友琼, 贾东芬, 等. 高效液相色谱法检测水产品中硝基呋喃类代谢物残留量[J]. 分析测试实验室, 2009(12): 86-90.
[7] 丁磊, 蒋俊树, 顾亮, 等. 高效液相色谱串联质谱法快速测定水产品中硝基呋喃类代谢物研究[J]. 现代农业科技, 2010(11): 336-339.
[8] ALEXANDER L, PETER Z, WOLFGANG L. Determination of the metabolites of nitrofuran antibiotics in animal tissue by highperformance liquid chromatography-tandem mass spectrometry[J]. Journal of Chromatography A, 2001, 939(1): 49-58.
[9] 杨琳, 傅红, 刘强, 等. 水产品及其苗种中硝基呋喃代谢物的高效液相色谱-串联质谱法测定[J]. 食品科学, 2010, 31(12): 206-211.
[10] 钱卓真, 位绍红, 余颖, 等. 高效液相色谱-串联质谱法测定鲍鱼中硝基呋喃类代谢物残留量[J]. 福建水产, 2010, 6(2): 43-49.
[11] JANE K F, JOSE L D, MAURO S, et al. Determination of nitrofuran metabolites in poultry muscle and eggs by liquid chromatographytandem mass spectrometry[J]. Journal of Chromatography B, 2005, 824(1): 30-35.
[12] 孙言春, 张洁, 许宪祝, 等. 固相萃取液相色谱串联质谱测定水产品中硝基呋喃类代谢物残留研究[J]. 中国渔业质量与标准, 2011, 2(1): 54-59.
[13] Commission Decision 93/256 /EEC. Analysis with dialysis and on-line HPLC for determining residues of nitromidazoles and nitrofurans in poultry (chickens, ducks, and turkeys) meat[S].
[14] 陈剑刚, 白艳玲, 梁素丹, 等. 固相萃取-液相色谱-串联质谱法测定水产品中硝基呋喃类代谢物[J]. 中国食品卫生杂志, 2013, 25(4): 338-343.
[15] 祝伟霞, 袁萍, 杨冀州, 等. 固相支撑液液萃取-平行蒸发前处理技术测定动物源性食品中四种硝基呋喃类代谢物[J]. 食品科技, 2011, 36(2): 300-303
[16] 农业部783号公告-1—2006水产品中硝基呋喃类代谢物残留量的测定液相色谱-串联质谱法[S].
[17] 农业部781号公告-4—2006动物源食品中硝基呋喃类代谢物残留量的测定高效液相色谱-串联质谱法[S].
[18] 于慧娟, 李冰, 蔡友琼, 等. 液相色谱-串联质谱法测定甲壳类水产品中氨基脲的含量[J]. 分析化学, 2012, 40(10): 1530-1535.
[19] 应永飞, 朱聪英, 陈慧华, 等. 多壁碳纳米管分散固相萃取结合LCMS/MS测定饲料中11种β-受体激动剂[J]. 中国畜牧杂志, 2013, 49(9): 68-73.
[20] 张毅, 岳振峰, 蓝芳, 等. 分散固相萃取净化与液相色谱/串联质谱法测定牛奶中8 类禁用药物残留[J]. 分析化学, 2012, 40(5): 724-729.
[21] 陈红平, 刘新, 王川丕, 等. 分散固相萃取-超高压液相色谱-串联质谱法测定茶叶中赤霉酸和α-萘乙酸[J]. 分析化学, 2012, 40(7): 1059-1064.
Determination of Nitrofuran Metabolite Residues in Aquatic Fingerlings by High Performance Liquid Chromatography-Mass Spectrometry with Dispersive Solid Phase Ex traction
YAN Zhong-yong1,2, ZHANG Xiao-jun1,2,*, MEI Guang-ming1,2, LI Pei-pei1,2, LONG Ju1,2, ZHU Yin1,2, WANG Jian3
(1. Marine Fisheries Research lnstitute of Zhejiang Province, Zhoushan 316021, China; 2. Key Laboratory of Sustainable Utilization of Technology Research for Fishery Resourceof Zhejiang Province, Zhoushan 316021, China; 3. College of Food Science and Biotechnology, Zhejiang Gongshang University, Hangzhou 310035, China)
A method has been developed for the determination of four metabolites of nitrofuran, i.e. semicarbazide (SEM), 3-amino-5-morpholinomethyl-2-oxazolidineone (AMOZ), 3-amino-2-oxazolidinone (AOZ) and 1-aminohydantoin hydrochloride (AHD) in aquatic fi ngerlings, by high performance liquid chromatography-mass spectrometry with dispersive solid phase extraction. Samples were washed with a mixture of methanol-water (8:1, V/V), subjected to HCl acidolysis and derivatized using 2-nitrobenzaldehyde. The derivative was extracted with ethyl acetate, then concentrated and cleaned up by dispersive solid phase extraction. The analysis was carried out in the multi-reaction monitoring (MRM) mode and internal standard isotope dilution method was used for quantifi cation. Good linearity was obtained in the correlations between the peak areas and concentrations of the four metabolites of nitrofuran antibiotics over the concentration range from 0.5 to 20.0 μg/L, with correlation coeffi cients above 0.997. The limits of detection (LODs) for the four compounds were all 0.1 μg/kg. The range of average recoveries was between 90.5% and 104.5%, with relative standard deviations (RSDs) between 1.56% and 8.08%. The effi cient and simple method could be used to identify and quantify the metabolites of nitrofuran antibiotics in aquatic fi ngerlings with satisfactory sensitivity and repeatability.
dispersive solid phase extraction; high performance liquid chromatography-mass spectrometry; aquatic fi ngerlings; nitrofuran metabolites
TS201.6
A
1002-6630(2014)16-0153-07
10.7506/spkx1002-6630-201416030
2013-10-22
浙江省自然科学基金项目(LQ13C200004);浙江省科技计划项目(2013C37072)
严忠雍(1990—),男,助理工程师,学士,研究方向为渔业环境监测和水产品质量安全。
E-mail:yzy123123123@126.com
*通信作者:张小军(1982—),男,工程师,博士,研究方向为水产品质量安全和标准化。E-mail:xiaojun3627@163.com
