内切葡聚糖酶基因在黑曲霉中的同源表达
2014-01-16李杰高博江连洲刘君邓晨旭陈璐璐张会
李杰,高博,江连洲,刘君,邓晨旭,陈璐璐,张会
(1.东北农业大学生命科学学院,哈尔滨 150030;2.黑龙江省高校农业生物功能基因重点实验室,哈尔滨 150030;3.东北农业大学食品学院,哈尔滨 150030)
内切葡聚糖酶基因在黑曲霉中的同源表达
李杰1,2,高博1,2,江连洲3,刘君1,2,邓晨旭1,2,陈璐璐1,2,张会1,2
(1.东北农业大学生命科学学院,哈尔滨 150030;2.黑龙江省高校农业生物功能基因重点实验室,哈尔滨 150030;3.东北农业大学食品学院,哈尔滨 150030)
内切葡聚糖酶作用于纤维素的非结晶区,随机水解β-1,4-糖苷键,将长链的纤维素截断,对纤维素的整体降解起重要作用。研究从黑曲霉CICC2462中扩增得到内切葡聚糖酶基因Eng1,并针对黑曲霉中高表达的糖化酶基因glaA位点,构建Eng1基因表达载体pSZHG-Eng1,进一步通过农杆菌介导法转化黑曲霉CICC2462。经潮霉素筛选和PCR鉴定获得2株在glaA基因位点发生基因置换的同源重组转化子。在摇瓶发酵条件下,发酵液上清中的内切葡聚糖酶活力最高可达到272 U·mL-1,是出发菌株5.2倍。SDS-PAGE分析显示,两株重组菌株都有约36 ku目的蛋白条带,表达量为165~193 μg·mL-1。结果表明,研究实现内切葡聚糖酶在黑曲霉中的同源表达,可为食品级内切葡聚糖酶的大规模工业化生产奠定基础。
内切葡聚糖酶;黑曲霉;同源重组;表达
纤维素酶是将纤维素降解为葡萄糖多组分酶系的总称,是最有应用潜力酶制剂之一。纤维素酶属于糖苷水解酶,传统上被分为3类组分:内切型-β-葡聚糖酶(endo-β-1,4-glucanase,EC 3.2.1.4),即CMC酶;外切型-β-葡聚糖酶(exo-β-l,4-glucanase,EC 3.2.1.91);β-葡萄糖苷酶(β-l,4-glucanase,EC 3.2.1.21)[1]。目前,生产纤维素酶主要依靠微生物发酵,微生物菌种大多为真菌,如木霉、青霉、曲霉及腐质霉等,被广泛用于生产纤维素酶。但纤维素酶菌株产量低、成本高及稳定性不理想等因素仍制约纤维素酶生产和应用。开展纤维素酶高产重组菌株及发酵工艺研究等工作尤为重要。
在纤维素酶系中,内切-β-葡聚糖酶是研究最广泛且最深入的酶。内切葡聚糖酶作用于纤维素长链分子内部,将长纤维切成短纤维。由于内切葡聚糖酶特异性作用于β-1,4-葡萄糖苷键,可部分酶解纤维。因此,在饲料、啤酒、纺织等工业中发挥重要作用[2-4],可分解大麦及麦芽中的β-葡聚糖凝胶,降低麦汁粘度,提高麦汁的过滤速度及得率;也可作为一种重要畜禽大麦饲料添加剂,大幅提高饲料生物转化率并减少排泄物对环境污染[5];在pH中性偏碱范围内,具有水解细小织物纤维能力,将其加入洗涤剂中可增强洗涤效果,对衣物还具有柔软、增艳作用。
黑曲霉自身能够分泌内切β-葡聚糖酶,但野生型菌株表达量低,含有外切型-β-葡聚糖酶、β-葡萄糖苷酶等组分,不能满足生产纤维低聚糖等特殊要求。本试验利用黑曲霉中高表达的糖化酶基因(glaA)位点的调控作用,实现内切β-葡聚糖酶在黑曲霉中的同源表达,为获得高产量、高纯度的内切β-葡聚糖酶奠定理论基础。
1 材料与方法
1.1 材料
1.1.1 试验材料
黑曲霉CICC2462由肇东市日成酶制剂有限公司提供;大肠杆菌DH5α、农杆菌AGL1来自东北农业大学生命科学学院遗传学研究室;质粒pSZHG由东北农业大学生命科学学院遗传学研究室构建。
1.1.2 试剂
Primer STAR酶、限制性内切酶、pMD18-T载体,T4DNA连接酶等均购自TaKaRa生物工程公司;DNA片段回收试剂盒购自天根生物工程公司;Hygromycin B购自美国Sigma公司;化学试剂均为国产分析纯。
1.2 方法
1.2.1 黑曲霉基因组的提取
采用CTAB法[6]提取黑曲霉CICC2462基因组。
8~9月香菇价格较高,但处黔西南州的高温季节,同期降雨量大,容易形成高温高湿的环境。在该季节主要注意降温工作,但最易被忽略的是棚内通风、换气,喷水后要及时通风0.5~1小时[5],高温高湿易导致出菇快、出菇薄且易开伞,这也是万家店片菇率高达15%的原因。在该季节为免烂棒、闷棒[6],脱袋转色优于袋内转色。温度升高、高温季节在山区可用脱袋转色,温度降低的季节采用袋内转色的方式管理菌棒。
1.2.2 引物设计
根据GenBank中公布的黑曲霉内切葡聚糖酶基因Eng1序列(序列号:XM_001391932.2),以及黑曲霉表达载体pSZHG多克隆位点的特征,用Prim⁃er 5.0软件分别设计Eng1基因引物P1、P2。根据黑曲霉表达载体pSZHG-Eng1的序列,以及黑曲霉糖化酶基因位点的序列设计转化子PCR检测引物P3、P4及同源重组转化子PCR检测引物P5、P6。引物序列如表1所示。

表1 本研究所用引物Table 1 Primers used in this research
1.2.3 基因扩增
以黑曲霉基因组为模板,利用基因特异引物,PCR扩增出内切葡聚糖酶Eng1基因片段。将PCR扩增出DNA产物加A回收,与pMD18-T载体连接,转化大肠杆菌感受态,挑取单菌落,提取质粒酶切鉴定,将酶切鉴定正确的转化子送交测序公司测序,得到测序结果后进行Blast比对,并分析序列信息。
1.2.4 黑曲霉表达载体的构建
用ApaⅠ和ClaⅠ双酶切鉴定正确的转化子质粒,回收1 227 bp的目的基因条带,再用相同的酶,酶切表达载体pSZHG,回收约为15 280 bp载体条带。将这两条具有黏性末端的片段利用T4DNA连接酶连接,构建黑曲霉表达载体pSZHGEng1,如图1所示。
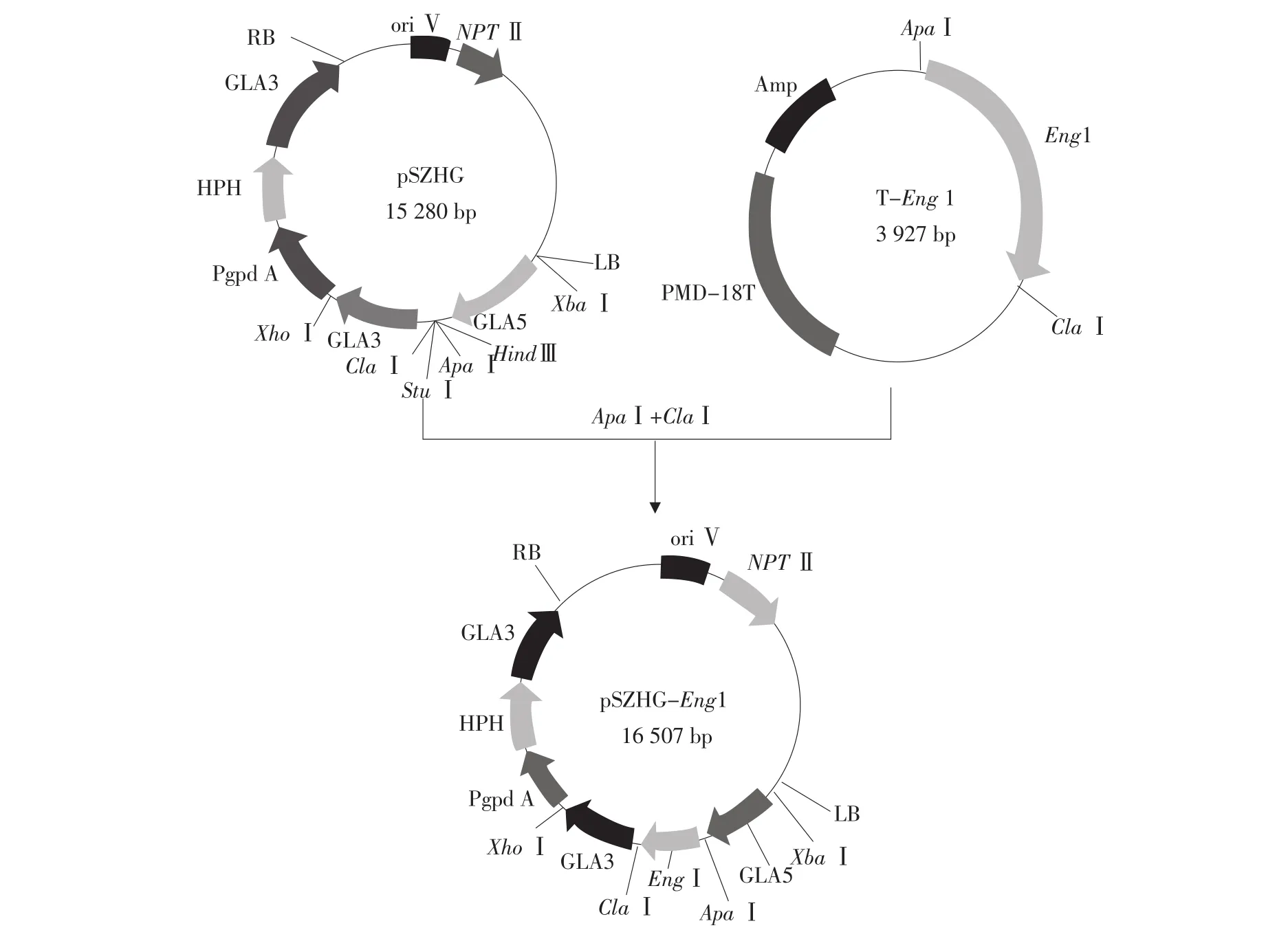
图1 pSZHG-Eng1的构建过程Fig.1 Construction of pSZHG-Eng1
1.2.5 黑曲霉的转化及同源重组菌株筛选
采用农杆菌介导法转化黑曲霉,采用PCR鉴定法筛选同源重组菌株[7]。
1.2.6 重组菌株酶活测定
取摇瓶发酵的内切葡聚糖酶发酵液上清,采用DNS法[8]测定内切葡聚糖酶酶活。
1.2.7 内切葡聚糖酶在重组菌株表达分析
取摇瓶发酵内切葡聚糖酶发酵液上清,12 000 r·min-1离心2 min,去尽上清,加入5×SDS-PAGE电泳上样缓冲液,沸水浴处理10 min,冷却至室温。然后,将培养液上清和细胞裂解物于浓度为10%SDS-PAGE凝胶进行凝胶电泳分析。
2 结果与分析
2.1 内切葡聚糖酶Eng1基因的扩增
以黑曲霉CICC2462基因组DNA为模板,利用P1、P2引物,PCR扩增出大小为1 227 bp的Eng1基因目的片段(见图2)。
将Eng1基因片段克隆、测序、进行Blast比对结果表明,克隆的Eng1基因与登录号XM_00139 1932.2基因序列相似性为99%。导致编码蛋白的第26位氨基酸由赖氨酸变为谷氨酸,经NCBI protein blast分析可知此位点不在蛋白质活性区域内,不影响蛋白质活性。
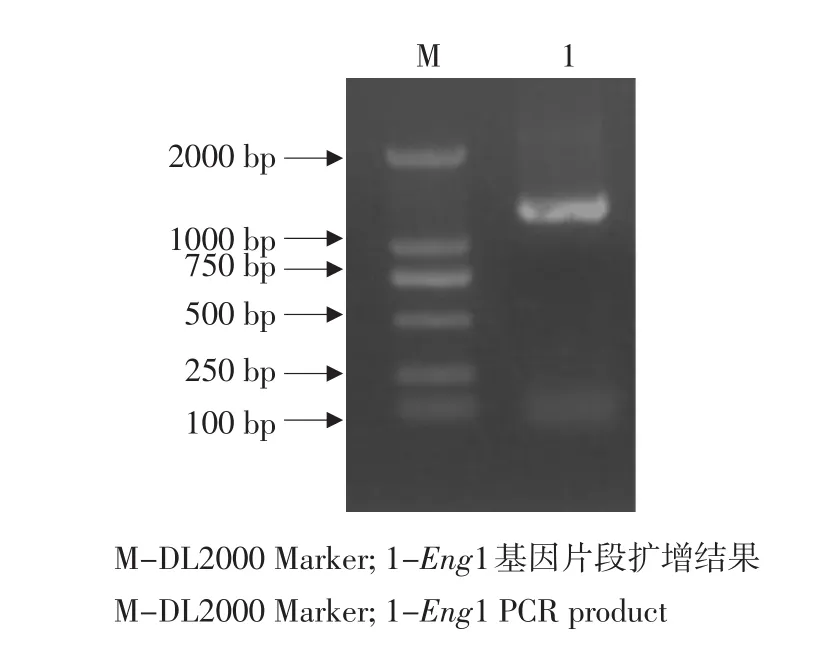
图2 Eng1基因片段PCR扩增结果Fig.2 Cloning of Eng1 by PCR
2.2 内切葡聚糖酶基因黑曲霉表达载体的构建
用ApaⅠ和ClaⅠ双酶切质粒pSZHG-Eng1,可切出1 227 bpEng1基因片段和15 280 bp pSZHG载体片段,说明载体构建正确(见图3)。

图3 pSZHG-Eng1表达载体的酶切鉴定结果Fig.3 Digestion of the expression vector pSZHG-Eng1 by the restriction enzyme
2.3 黑曲霉转化子的筛选和鉴定
将农杆菌转化子与黑曲霉在筛选培养基上共培养6~8 d,可见明显生长的黑曲霉菌落,挑取筛选平板上的单菌落于液体培养基中继续培养,直至长出菌丝体,提取黑曲霉菌丝体基因组DNA,利用P3、P4引物PCR鉴定,结果筛选出2株阳性转化子(见图4)。
2.4 同源重组转化子的筛选与鉴定
对筛选出的2株阳性转化子进行同源重组筛选,利用P5、P6引物对其进行PCR鉴定,结果2株转化子均扩增出1 701 bp目的带(见图5),可知均为同源重组转化子,同源重组转化率为100%。

图4 pSZHG-Eng1重组转化子PCR鉴定结果Fig.4 Identification of recombinants containing pSZHG-Eng1 by PCR

图5 同源重组转化子PCR鉴定结果Fig.5 Results of PCR of homologous recombination transformants
2.5 重组菌株酶活的测定
利用发酵培养基摇瓶培养内切葡聚糖酶重组菌株,从第5天开始取样,连续取9 d,离心取发酵液上清,进行酶活测定。其中第11天酶活最高达272 U·mL-1,是出发菌株的5.2倍。测定结果见图6。
2.6 内切葡聚糖酶在重组菌株中的表达分析
根据酶活分析,取摇瓶发酵培养11 d的发酵液上清样品进行SDS-PAGE分析(见图7),可见,2株重组菌株都有约36 ku目的蛋白条带,经AL⁃PHAIMAGER SYSTEMS软件分析,表达量为165~193 μg·mL-1,而出发菌株仅为38 μg·mL-1。
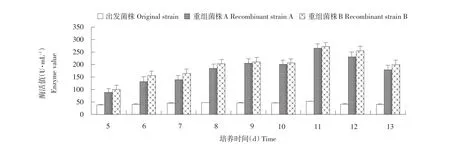
图6 不同培养时间发酵液上清中的内切葡聚糖酶酶活Fig.6 Enzyme activity detection at different time of Eng1
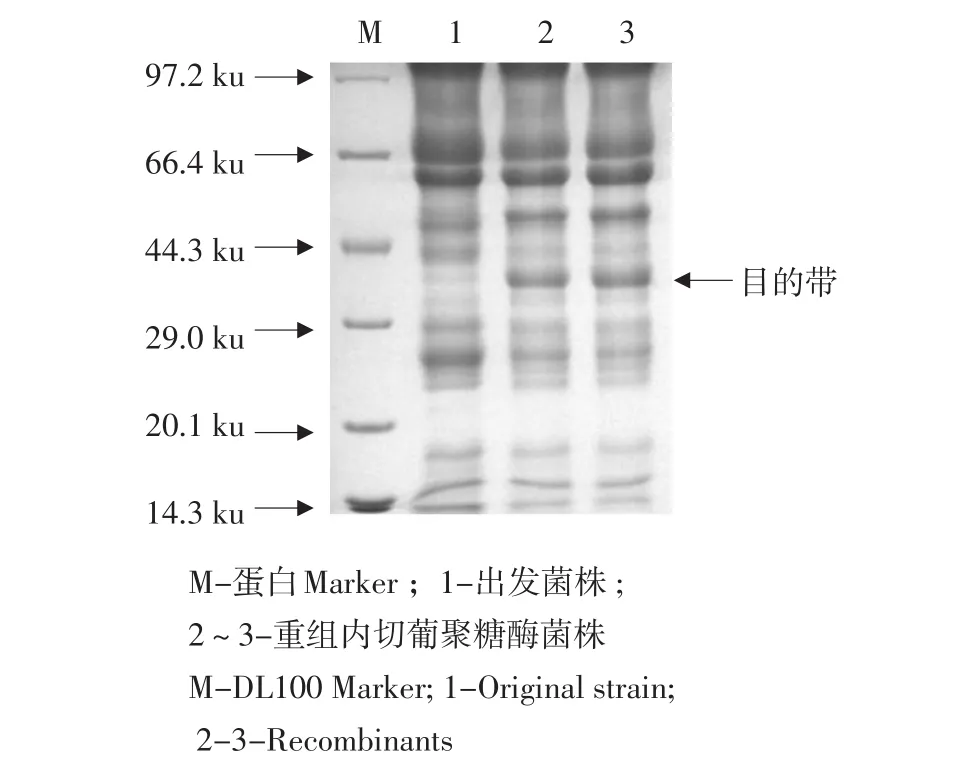
图7 SDS-PAGE蛋白表达凝胶电泳图谱Fig.7 SDS-PAGE profile of the expressed protein
3 讨论与结论
近年来关于内切葡聚糖酶基因克隆与表达研究,常见宿主菌有大肠杆菌、毕赤酵母、丝状真菌等。官兴颖等克隆得到枯草芽孢杆菌C-36的内切葡聚糖酶基因,以大肠杆菌为宿主进行融合表达,测得产物CMC酶活力为5.94 U·mL-1[9]。陈惠等将枯草芽孢杆菌的内切葡聚糖酶基因在巨大芽胞杆菌中进行外源表达,工程菌大罐发酵可检测到酶活为889 U·mL-1[10],但利用巨大芽胞杆菌表达发现表达内切葡聚糖酶对宿主菌具有毒性,严重影响工程菌生长[11]。Rubini等克隆Penicillium echinu⁃latum的内切葡聚糖酶基因EGI,在毕赤酵母中实现表达,EGI酶的最适条件为pH 5.0~9.0,同时具有良好耐热性[12]。Mala等分别将来自里氏木霉、哈次木霉及绿色木霉的内切葡聚糖酶基因导入酿酒酵母中进行表达,表达产物中,来自里氏木霉的葡聚糖内切酶的酶活是出发菌株3倍,来自哈次木霉和绿色木霉内切葡聚糖酶酶活均提高至出发菌株的2倍[13]。顾斌涛等成功实现特异腐质霉中的中性内切-β-葡聚糖酶在里氏木霉中重组与胞外表达,其摇瓶发酵酶活达到98.8 U·mL-1[14]。黑曲霉是世界公认安全生产菌种,也是内切葡聚糖酶主要的宿主菌之一。同时黑曲霉优良的蛋白质分泌能力、准确的翻译后加工、与高等真核生物类似等特点被广泛应用于工业酶制剂加工受体菌中[15]。本试验通过克隆得到基因Eng1,使其在黑曲霉中进行同源表达,内切葡聚糖酶酶活最高达272 U·mL-1,这与在毕赤酵母中表达量(1 928 U·mL-1)相比尚有差距[16],本研究可进一步筛选纯合同源重组转化子以提高优化菌株目的蛋白表达量。经Alphaimager Systems软件计算,目的蛋白占菌株总蛋白表达量1/10,说明目的蛋白表达量相对较高,可通过对发酵条件进一步优化,提高内切葡聚糖酶表达。
本研究实现内切葡聚糖酶在黑曲霉中同源表达,获得2株重组菌株的目的蛋白表达量达到165~193 μg·mL-1,表达量较高,可为内切葡聚糖酶工业化生产奠定基础。经Alphaimager Systems软件分析SDS-PAGE中目的蛋白相对分子质量约为36,比内切葡聚糖酶理论分子质量33.7 ku稍大。通过NetNGlyc 1.0 Server软件分析发现内切葡聚糖酶氨基酸序列中可知有2个糖基化位点,该位点糖基化导致重组蛋白实际分子质量高于理论值。
[1]方诩,曲音波.纤维素酶在木质纤维素转化产糖技术中的开发与应用[J].生物产业技术,2010(4):48-51.
[2]Sukumaran R K,Singhania R R,Mathew G M,et al.Cellulase pro-duction using biomass feed stock and its application in ligno⁃cellulose saccharification for bio-ethanol production[J].Renew⁃able Energy,2009,34(2):421-424.
[3]Gowen C M,Fong S S.Exploring biodiversity for cellulosic biofuel production[J].Chemistry&Biodiversity,2010,7(5):1086-1097.
[4]Takashima S,Ohno M,Hidaka M,et al.Correlation between cel⁃lulose binding and activity of cellulose-binding domain mutants of Humicola grisea ellobiohydrolase[J].FEBS Letters,2007,581 (30):5891-5896.
[5]岳淑宁,张红艳,张强,等.饲用纤维素酶质量评价的研究[J].中国酿造,2009(1):153-154.
[6]王凡,洪葵.CTAB法提取野野村菌基因组DNA[J].微生物学通报,2010,37(8):1212.
[7]李杰,张会,张莹莹,等.食品级木聚糖酶黑曲霉工程菌的构建[J].东北农业大学学报,2013,44(11):7-13.
[8]郑海英,黄平,蔡少丽,等.特异腐质霉内切葡聚糖酶且基因在毕赤酵母中的表达及酶学性质[J].微生物学通报,2012,39 (2):145-153.
[9]官兴颖,吴振芳,吴琦,等.枯草芽孢杆菌C-36内切葡聚糖酶基因的克隆及其在大肠杆菌中融合表达[J].生物加工过程,2009, 7(3):68-72.
[10]陈惠,青兵,廖俊华,等.内切葡聚糖酶基因在巨大芽胞杆菌中的表达及其酶学性质研究[J].遗传,2008,30(5):649-654.
[11]吴振芳,陈惠,曾民,等.内切葡聚糖酶基因在毕赤酵母中高效表达研究[J].农业生物技术学报,2009,17(3):529-535.
[12]Rubini M R,Dillon A J P,Kyawl C M,et al.Cloning of character⁃ization and heterologous expression of the first penicillium echinu⁃latum cellulase gene[J].Journal of Applied Microbiology,2010, 108(4):1187-1198.
[13]Mala C G,Sumangala B,Pranav C,et al.Cloning and expression of endo-glucanase genes from Trichoderma species in Saccharo⁃myces cerevisiae[J].Journal of Applied Sciences Research,2008,4 (11):1546-1556.
[14]顾斌涛,江守坤,夏黎明.中性内切-β-葡聚糖酶基因在里氏木霉中的重组与表达[J].高校化学工程学报,2013,27(1):108-112.
[15]郭艳梅,郑平,孙际宾.黑曲霉作为细胞工厂:知识准备与技术基础[J].生物工程学报,2010,26(10):1410-1418.
[16]黄君,张昌毅,梁运祥,等.黑曲霉β-1,4-葡聚糖酶在毕赤酵母中的高效表达[J].华中农业大学学报,2008,27(5):611-615.
Endo-glucanase gene homologous expressed inAspergillus niger
LI Jie1,2, GAO Bo1,2,JIANG Lianzhou3,LIU Jun1,2,DENG Chenxu1,2,CHEN Lulu1,2,ZHANG Hui1,2(1.School of Life Sciences,Northeast Agricultural University,Harbin 150030,China;2.Key Laboratory of Agricultural Biological Functional Gene of Heilongjiang Province,Harbin 150030,China;3.School of Food Science, NortheastAgricultural University,Harbin 150030,China)
An endo-glucanase can truncate long chains of cellulose in cellulose non-crystalline region through random hydrolysis ofβ-1,4-glycosidic linkage,which plays an important role in the overall degradation of cellulose.In order to improve the endo-glucanase activity ofAspergillus niger,a gene designatedEng1 encoding an endo-glucanase fromAspergillus nigerCICC2462 was cloned by using the reverse transcription PCR method.Using the highly expressed elment ofglaAencoding a glucoamylase,the recombinant expression vector pSZHG-Eng1 was constructed and transformed into the strain CICC2462 via the Agrobacterium-mediated method.Two strains were obtained by screening hygromycin resistant mutants and base pair substitution based on homologous recombination was established by PCR to exist inglaA gene loci gene.Under the shaking flask fermentation condition,the endo-glucanase activity of theA.niger transformant reached 272 U·mL-1in the fermentation solution,which was 5.2-fold as high as that of the original strain.The approximately 36 ku protein band from two strains was observed on the SDS-PAGE,and the expressed enzyme was quantitated to be 165-193 μg·mL-1.This results showed that the endo-glucanase gene was successfully transformed and homologously expressed inAspergillus niger,which laid thefoundation for the construction of an engineered strain to produce a food-grade endo-glucanase in large-scale industrial production.
endo-glucanase;Aspergillus niger;homologous recombination;expression
TQ925;Q786
A
1005-9369(2014)09-0056-06
2014-02-22
国家863高技术研究发展计划(2013AA102104)
李杰(1972-),男,副教授,博士,研究方向为分子遗传学与基因工程。E-mail:lijie_neau@126.com
时间2014-9-18 10:50:03[URL]http://www.cnki.net/kcms/detail/23.1391.S.20140918.1050.015.html
李杰,高博,江连洲,等.内切葡聚糖酶基因在黑曲霉中的同源表达[J].东北农业大学学报,2014,45(9):56-61.
Li Jie,Gao Bo,Jiang Lianzhou,et al.Endo-glucanase gene homologous expressed inAspergillus niger[J].Journal of Northeast Agricultural University,2014,45(9):56-61.(in Chinese with English abstract)
