具有强可见光吸收的富勒烯-Bodipy衍生物作为光催化剂在硫醚光催化氧化中的应用
2014-01-05赵建章
黄 灵,赵建章
(大连理工大学 化工学院 精细化工国家重点实验室,辽宁 大连116024)
三 重 态 光 敏 剂 在 光 催 化 有 机 反 应[1-6]、光 聚合[7-9]、光动 力 治 疗[10-13]以 及 三 重 态-三 重 态 湮 灭(TTA)上转换等领域[14-16],都具有重要的应用。目前在光催化有机合成反应中应用的大多数光敏剂为Ru(Ⅱ)、Ir(Ⅲ)、Pt(Ⅱ)等贵金属配合物[17-20],这些光催化剂存在一些局限,例如可见光吸收能力弱(ε<10000mol-1·L·cm-1),三重激发态寿命较短 (τT<10μs),这些因素导致催化剂和底物间的分子间能量转移或电子转移效率较低,所以导致所需的光催化反应时间延长,并且催化剂价格昂贵。虽然含碘、溴原子的荧光素类染料(如孟加拉玫瑰红、曙红Y)也被应用于光催化有 机 合 成 反 应[21,22],但 是 这 类 光 敏 剂 的 分子结构难以衍生化以改善其吸光能力和氧化还原电位。在光催化有机合成反应中,三重态光敏剂(光催化剂)一般是做为电子的受体或给体,与反应底物间发生单电子转移反应,属分子间反应,所以处于三重激发态的光催化剂的浓度(与催化剂的吸光能力成正比)越高、光催化剂三重态的寿命越长,就越有利于提高分子间的电子转移效率,从而实现高效的光催化反应。因此有必要开发具有分子结构可调、具有强可见光吸收、长三重激发态寿命的催化效率更高的有机三重态光敏剂。
氟硼吡咯类染料 (Bodipy)光稳定性良好,荧光量子产率高,分子结构易于衍生化,因此受到了广泛的关注[23-26]。但是这类染料的三重态量子效率低,不适于做为三重态光敏剂。为了提高其三重态量子效率,通常采用重原子效应[27,28]或者三重态能量转移(敏化)的方法[29-31],使其三重激发态得到布居。而富勒烯(C60)具有高的三重态量子效率,但是其可见光吸收能力较弱,因此也不适于用作三重态光敏剂。
通过共价键将Bodipy和C60连接起来,可以综合二者的优点,通过能量转移、利用能量受体C60的自旋转换单元的作用[16],可以制备出具有强可见光吸收、高效系间窜越(高三重态量子产率)且不含重原子的三重态光敏剂[29-31]。本文利用该类新型有机三重态光敏剂做为光催化剂,以空气中的分子氧(O2)作为氧化剂进行了光催化氧化硫醚的反应。利用分子氧的氧化反应是绿色化学研究的主要内容[32],但是基态的分子氧氧化能力较弱,为了增加分子氧的氧化能力,可通过光敏化将其转变为单线态氧(1O2)。光敏剂通过激发态能量转移,或电子转移产生单线态氧或超氧负离子自由基(O2·-)[33]。单线态氧和超氧负离子自由基可以高选择性地氧化硫醚[34-36],但目前所报道的光敏剂存在可见光吸收较弱、所需反应时间较长和光敏剂稳定性差等缺点[36]。
本文通过Suzuki反应和Proto反应,制备了具有可见光强吸收的 C60-Bodipy Dyad 和 C60-Bodipy Triad衍生物(见图1)。将其应用于光催化氧化硫醚反应,与传统的Ru(Ⅱ)、Ir(Ⅲ)配合物以及曙红等光敏剂相比,三重态寿命长达92.1 μs,光催化效率大大提高,反应时间得到大幅缩短,表明该类光敏剂是一类性能优良的光催化剂,有望在光催化有机合成反应中得到广泛的应用。
1 实验部分
1.1 试剂和仪器
实验中所采用的硫醚和相关的衍生物为阿拉丁化学试剂公司的产品,富勒烯购于南京先丰纳米材料公司。甲苯用分子筛干燥。光谱测试所用试剂均为色谱纯。
荧光光谱和紫外可见吸收光谱分别使用Shimadzu RF5301荧光光谱仪和 Agilent 8453紫外可见吸收光谱仪测定。荧光寿命和三重激发态寿命分别利用OB920荧光寿命仪(Edinburgh,UK)和LP920激光闪光光解仪(Edinburgh,UK)测定。1HNMR和电子顺磁共振(EPR)分别利用400 MHz Bruker核磁共振仪和 Bruker ESP-300E 电子顺磁共振仪测定。
1.2 光氧化实验步骤
甲基苯基硫醚(0.2mmol,25mg)和光敏剂(1.2mg,0.5mol%)加入到10mL的单口瓶中,再加入3mL CH2Cl2/CH3OH (9/1,体积比)作为溶剂,利用35W 氙灯做激发光源,用0.72 mol/L的NaNO2溶液过滤除去波长短于380nm的紫外光。光强调整到50mW/cm2。TLC检测反应进程,当底物完全转化以后,减压除去溶剂,利用核磁共振氢谱确定收率。其中底物的特征峰为2.40(甲基上的质子),产物的特征峰为2.72.
1.3 利用 EPR 检测单线态氧(1 O2)和超氧负离子自由基(O2-·)
以2,2,6,6-四甲基哌啶(TEMP)作为单线态氧捕获剂,5,5-二甲基氮氧化物(DMPO)作为超氧负离子自由基捕获剂;其产物是稳定的自由基,能够被EPR检测到。其中,TEMP和DMPO所用浓度分别为0.1mol/L和0.001mol/L。敏化剂浓度为1.0×10-5mol/L,甲基苯基硫醚的浓度为3.0×10-3mol/L,所用的激光器波长为532nm(二极管泵浦固体连续激光器),激光光束功率密度为141mW/cm-2。将上述化合物混和以后放入到直径为2mm、长度为3cm的石英管中进行测定。
1.4 敏化剂光物理性质测定
将敏化剂配制为1.0×10-3mol/L的溶液,测试光谱时稀释到1.0×10-5mol/L。其中单线态氧量子效率以孟加拉玫瑰红 (单线态氧量子产率ΦΔ=0.80,CH3OH)和亚甲基蓝(ΦΔ=0.57,CH2Cl2)做为标准物质进行测定。
单线态氧量子产率的计算公式如下:
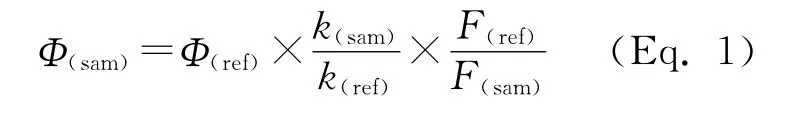
其中,Φ(ref)为参比化合物的单线态量子效率,k(sam)和k(ref)分别为氧化1,3-二苯基苯并呋喃时在414 nm处斜率的变化,F(ref)和F(sam)分别为激发光处吸光度矫正因子(吸收光子数的相对值)。

图1 敏化剂B-1、B-2和B-3以及对照化合物的分子结构
2 结果与讨论
2.1 敏化剂的光物理性质
从图2可以看出,B-1和B-2在517nm和630 nm 具有强的可 见光吸收能 力[29,31]。B-3具有 宽谱带吸收的特点(480nm 到650nm)[30]。其中,515nm处的吸收峰来源于2-苯基Bodipy部分,630nm处的吸收峰来源于Distyryl-Bodipy部分。与之相比,传统的光敏剂Ru-1(452nm,ε=13000 mol-1·L·cm-1)、Ir-1 (255nm,ε=11300 mol-1·L·cm-1)、曙红 Y(536nm,ε=42000 mol-1·L·cm-1)在可见区的吸光能力是比较弱的。与对照化合物B-4、B-5和B-6相比,敏化剂B-1~B-3在可见区的强吸收主要是来源于光吸收天线Bodipy 部分[29-31]。由 于 B-3的荧光激 发 光谱和吸收光谱具有很好的重合性,说明从2-苯基Bodipy部分到二苯乙烯基-Bodipy具有高效的能量转移效率(95%)[31]。

图2 (a)B-1、B-2和B-4、B-5的紫外可见吸收光谱;(b)Ru-1、Ir-1和曙红 Y的紫外可见吸收光谱;(c)B-3和B-6的紫外可见吸收光谱;(d)归一化的B-3的激发光谱和吸收光谱,c=1.0×10-5 mol/L,溶剂为二氯甲烷

表1 光敏剂与相关的对照化合物的光物理性质aPhotophysical parameters of the photosensitizers and the reference compoundsa
从表1可以看出,B-1~B-3具有低的荧光量子效率,这主要是因为发生了从Bodipy部分到富勒烯部分的能量转移,从而使其荧光淬灭[29-31]。通过纳秒时间分辨瞬态吸收光谱,测得B-1~B-3的三重态寿命分别为27.4μs、71.2μs和92.1 μs。同时B-1~B-3还具有较高的单线态氧量子效率(ΦΔ,表1)。对于B-3,分别采用515nm 和624nm的单色光进行激发,测得的单线态氧量子效率分别为0.67和 0.90。与之相比,Ru-1和Ir-1的三重态寿命较短、单线态氧量子效率较低(表1),虽然曙红Y在可见区具有较强的吸收,但是其三重态寿命 (3.5μs)和单线态氧量子效率 ((ΦΔ=0.31)很低,不利于提高光催化反应中催化剂和底物的分子间能量转移或电子转移的效率。
2.2 光催化氧化硫醚
为了获得最佳的光催化硫醚氧化反应效果,分别对反应时间、照射光强、敏化剂用量和反应气氛等反应条件进行了优化,发现在空气中、光强为50mW/cm2、以 B-1敏化剂、反应时间为20min时就可以获得满意的收率(表2)。而使用Ru-1,Ir-1或曙红Y,即使将反应时间延长3倍也没有将底物完全转化(表2)。这充分说明具有强可见光吸收的C60-Bodipy dyad是一种性能优良的光氧化硫醚的光敏剂。


表2 光氧化硫醚实验条件的优化Optimization of experimental conditions for photooxidation of thioanisolea
为了研究光敏剂对底物的适应性,将不同结构的硫醚衍生物也用于光催化氧化反应(表3)。研究发现,使用溴代、氯代的吸电子基团的硫醚,发现反应速率有所降低,但是对收率没有影响。敏化剂对于含有苄剂的硫醚也具有很好的光催化氧化效果;三元环具有很大的张力,为了确定三元环在硫醚光氧化实验中是否稳定,将含有三元环的硫醚用于光催化氧化,发现三元环对反应没有影响,没有其它开环产物的生成。但是对于含有硝基的硫醚,没有取得良好的光氧化效果,可能是因为硝基化合物对于单线态氧具有淬灭效果[34]。
具有宽谱带吸收 的 光 敏 剂 B-3 (C60-Bodipy triad)也被用于光催化氧化反应,与只含有单发色团的B-1、B-2相比,反应时间大大缩短,例如对于甲基苯基硫醚,反应仅用11min就可完成。
为了进一步研究具有宽谱带吸收的光敏剂B-3具有较强的光催化氧化硫醚能力的原因,采用滤光片得到了470nm~520nm的光,用于光催化反应。从紫外-可见吸收光谱可以得知,对于B-3,这一吸收范围为2-苯基Bodipy部分的吸收,B-1主要的吸收的吸收范围也是470nm~520nm,而B-2在这一波长范围内没有显著的吸收。在这一波段下进行光氧化硫醚的反应,与B-2相比,发现利用B-1和B-3得到了较好的收率(表4)。这充分说明,对于具有宽谱吸收特点的B-3,通过能量转移有效的增加了光氧化硫醚的能力。

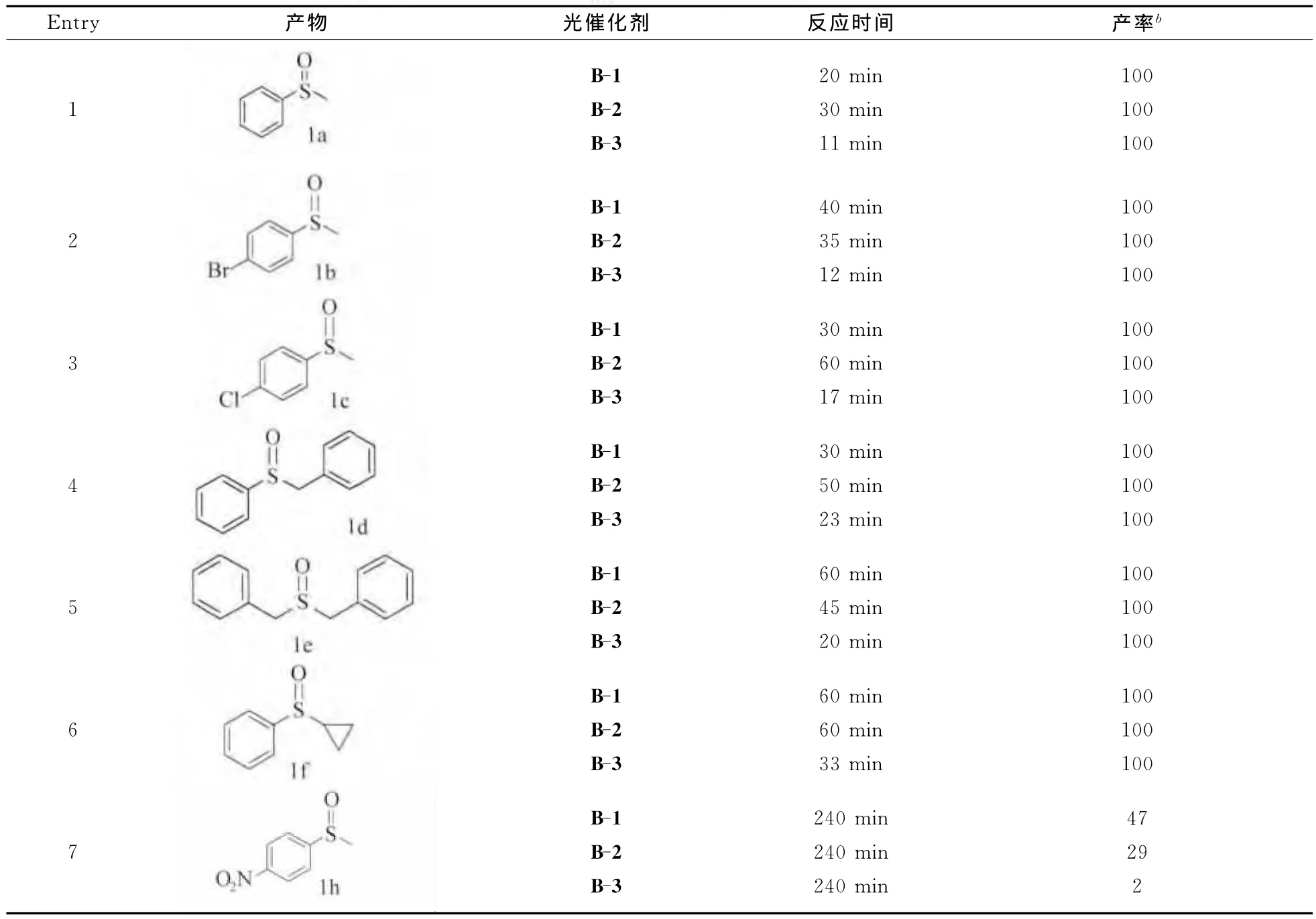
表3 光敏剂B-1,B-2和B-3用于硫醚衍生物的光催化氧化效果Visible light promoted aerobic photocatalytic oxidation of various sulfides with B-1,B-2 and B-3 as photocatalystsa


表4 B-1,B-2和B-3光催化氧化硫醚能力的比较Comparison of the photocatalytic ability of B-1,B-2 and B-3 for photooxidation for thioanisolea
硫醚是一种电子给体,以硫醚作为电子给体,光敏剂作为电子受体,其中光敏剂的T1态能级作为E0,0值。对于B-1三重态布居于C60,三重态能级为1.52eV。对于 B-2和 B-3三重态布居于Distyryl-Bodipy部分,三重态能级为1.21eV。对于B-1~B-3,从硫醚到光敏剂的电子转移过程的具有较大的驱动力,相应的吉布斯自由能变化分别为-0.74eV、-0.59eV、-0.50eV。当敏化剂接受电子以后,可以将电子转移到基态的分子氧,形成超氧负离子自由基(O2·-),超氧负离子自由基能够加速硫醚的光催化氧化反应[35,36]。
为了进一步探索光氧化硫醚的活性氧物种,采用化学淬灭的方法对反应机理进行了研究。由于氘代溶剂与普通溶剂相比,能够有效的稳定单线态氧,所以采用氘代的溶剂可以加速以单线态氧为活性氧化物种的光催化氧化反应[36]。苯醌是一种良好的超氧负离子自由基的捕获剂,能够快速的清除反应体系内的超氧负离子自由基[37],所以向反应体系中加入苯醌,能够抑制超氧负离子自由基参与的化学反应,能够有效的降低化学反应速率。通过比较表6中的第1和2项,发现在氘代的溶剂中,反应收率提高比较明显。对于单线态氧参与的硫醚光催化氧化反应,由于反应的中间体存在电荷,在含有质子性的极性溶剂中可以有效的稳定反应过程中形成的中间体,从而加速反应速率[38]。表6中的第3项表明,没有加入质子性的极性溶剂——甲醇,发现反应速率大大的降低。说明单线态氧参与了以C60-Bodipy作为光敏剂的硫醚氧化反应。为了进一步研究超氧负离子自由基对反应的影响,本文利用苯醌作为超氧负离子自由基的捕获剂用于光氧化硫醚反应。通过比较表6中4和5发现,加入苯醌以后反应速率大大的降低,这进一步说明,超氧负离子自由基也参与了光氧化硫醚反应。
表5 光敏剂B-1~B-3的氧化还原电位和分子间电荷转移自由能的计算Redox potentials(ERed and EOx)of Bodipy photocatalysts and the calculated free-energy changes for the charge separationΔ

表5 光敏剂B-1~B-3的氧化还原电位和分子间电荷转移自由能的计算Redox potentials(ERed and EOx)of Bodipy photocatalysts and the calculated free-energy changes for the charge separationΔ
a循环伏安图在除氧的二氯甲烷中测定。支持电解质[Bu4N][PF6]的浓度为0.10mol/L,光敏剂的浓度为1.0×10-3 mol/L,二茂铁作为内标物,对电极为铂电极,工作电极为玻碳电极,Ag/AgNO3作为参比电极.bΔGeT=e[Eox-Ered]-E0,0+ΔGs,光诱导电子转移(硫醚做为电子给体,光敏剂做为电子受体),其中Eox为硫醚的氧化电位,Ered为光敏剂的还原电位,E0,0是光敏剂的T1能级,对于B-1,以C60的三重态能级1.53eV进行计算,对于B-2和B-3,以Distyryl-Bodipy的三重态能级1.21eV进行计算.在极性溶剂中,ΔGs可忽略不计。aCyclic voltammetry was measured in N2saturated CH2Cl2containing a 0.10mol/L [Bu4N][PF6]supporting electrolyte;counter electrode is Pt(Ⅱ)electrode;working electrode is glassy carbon electrode;Ag/AgNO3couple as the reference electrode.25℃.Conditions:1.0×10-3 mol/L photocatalysts and 0.5×10-3 mol/L ferrocene in CH2Cl2.bΔGeT=e[Eox- Ered]- E0,0+ΔGs.Photoinduced electron transfer from thioanisole to photocatalysts,E0,0is the energy level of T1photocatalysts for B-1,the triplet state level is 1.52eV,for B-2 and B-3,the triplet state level is 1.21eV.In polar solvent theΔGscan be neglected.
ERed EOx ΔGeT b+0.012 --0.63 - -B-1 -0.78 +1.21 -0.74 B-2 -0.61 +1.23 -0.59 B-3 -0.69 +0.74 -0.50 Thioanisole C60-

表6 不同实验条件下B-1作为光敏剂的硫醚光氧化反应Visible light promoted aerobic oxidation of sulfide with B-1in different conditiona
电子自旋共振(ESR)是一种有效地检测自由基的实验手段。TEMP能够很好的捕获单线态氧,形成具有电子顺磁共振信号的稳定自由基。当体系存在单线态氧时,TEMP与单线态氧的加和物可以给出单线态氧存在的证据[39]。DMPO是一种有效的超氧负离子自由基捕获剂,能够与光催化反应体系中的超氧负离子自由基结合,形成具有电子顺磁共振信号的稳定自由基[39]。以532nm激光为激发光源对光敏剂与DMPO的混合物进行光照,没有观察到超氧负离子自由基与DMPO加合物的信号(图4a),说明在无反应底物存在下,混合物中不存在超氧负离子自由基;但是当光敏剂与TEMP混合以后,观察到了强的单线态氧信号(图4c),这说明光敏剂在光激发下有单线态氧产生。当在光敏剂与DMPO的混合溶液中加入反应底物甲基苯基硫醚,在532nm激光激发下,观察到了超氧负离子自由基与DMPO加合物的信号(图4d),而相应的单线态氧信号发生了减弱。该结果说明在甲基苯基硫醚存在下(做为电子给体,光催化剂做为电子受体),能够产生超氧负离子自由基。
图5给出了利用B-1,B-2和B-3光催化氧化硫醚的机理。当敏化剂被可见光激发以后,通过光收集天线向C60的单重态能量转移,经过C60的系间窜越,到达三重激发态(三重态定域于C60,或是苯乙烯基Bodipy基团上);处于三重激发态的敏化剂接受硫醚给出的电子(分子间电子转移),敏化剂在将电子转移给分子氧,形成超氧负离子自由基,从而进行硫醚的氧化。处于三重激发态的敏化剂也可以通过分子间三重态能量转移的方式,将三重态能量转移给基态氧,形成单线态氧,从而进行硫醚的氧化。由于反应体系中存在两种反应途径,同时B-1~B-3的可见光吸收能力很强,所以大大加快了光催化氧化硫醚的反应速率。
为了验证该光催化氧化硫醚是否具备实际制备应用价值,将光氧化硫醚的反应放大到10 mmol(1.24g甲基苯基硫醚),反应的收率和选择性并没有因为底物量的增加而降低 (催化剂B-1的用量:0.5mol%;光照强度:50mW/cm2;反应时间40min,1HNMR 产 率:100%)。该 结 果 说明,本文制备的新型三重态光敏剂在光氧化硫醚方面具有较高的实用价值。该类新型有机光催化剂也有望用于其他光催化有机反应中。
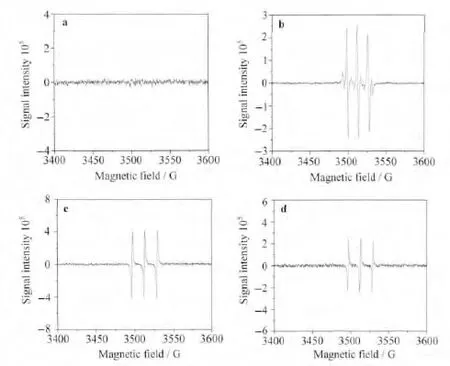
图4 光催化硫醚氧化反应机理的研究.不同混合物经光激发后的电子自旋共振光谱
3 结论
为了解决目前光催化有机合成反应中使用的光催化剂多为可见光吸收能力弱、分子结构不易修饰以及含贵金属(或重原子)等问题,本文根据氟硼吡咯(Bodipy)的强可见光吸收性质、以及富勒烯C60的高效系间窜越能力,将二者共价连接在一起,以使分子内共振能量转移过程能够发生,制备了二元以及三元化合物(Dyad以及Triad),做为不含任何重原子、具有宽谱带可见光吸收能力的有机三重态光敏剂(三重态寿命长达92.1μs),并将该类有机三重态光敏剂用于光催化硫醚氧化反应。研究发现,与传统的光敏剂Ru(Ⅱ)配合物、曙红Y的催化效果相比,新光敏剂对硫醚光氧化反应的催化效果有了显著提高,反应时间由传统催化剂的1h缩短至11min,并且反应的收率和选择性均接近100%。通过将具有分子内能量转移特点的C60-Bodipy的Dyad和Triad首次应用于光氧化硫醚,验证了通过分子内能量转移机理获得的宽谱带可见光吸收,可以进一步促进光敏剂光氧化能力。通过循环伏安法、电子转移自由能的计算、电子顺磁共振谱证明单线态氧以及超氧负离子自由基的生成等方法,详细研究了反应的机理。发现在光催化氧化硫醚反应过程中,超氧负离子自由基和单线态氧同时起作用,从而加快了硫醚的光催化氧化的反应速率。该类新型无重原子有机三重态光敏剂具有强的宽谱带可见光吸收能力、吸收波长灵活可调等优点。对具有长寿命三重激发态的有机三重态光敏剂的研究,将对新型三重态光敏剂的分子设计以及在光催化有机合成反应中的应用,起到一定的促进作用。
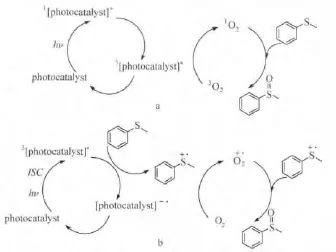
图5 C60-Bodipy Dyad以及C60-Bodipy Triad作为光敏剂光氧化硫醚的反应机理
[1] Xuan J,Xiao W J.Visible-light photoredox catalysis[J].Angewandte Chemie International Edition,2012,51(28):6828-6838.
[2] Narayanam J M R,Stephenson C R J.Visible light photoredox catalysis:applications in organic synthesis[J].Chemical Society Reviews,2011,40(1):102-113.
[3] Shi L,Xia W.Photoredox functionalization of C—H bonds adjacent to a nitrogen atom [J].Chemical Society Reviews,2012,41(23):7687-7697.
[4] Xi Y,Yi H,Lei A.Synthetic applications of photoredox catalysis with visible light [J].Organic & Biomolecular Chemistry,2013,11(15):2387-2403.
[5] Prier C K,Rankic D A,MacMillan D W C.Visible light photoredox catalysis with transition metal complexes:appli-cations in organic synthesis[J].Chemical Reviews,2013,113(7):5322-5363.
[6] Tucker J W,Stephenson C R J.Shining light on photoredox catalysis:theory and synthetic applications[J].Journal of Organic Chemistry,2012,77(4):1617-1622.
[7] Lalevée J,Peter M,Dumur F,Gigmes D,Blanchard N,Tehfe M A,Morlet-Savary F,Fouassier J P.Subtle Ligand effects in oxidative photocatalysis with iridium complexes:application to photopolymerization[J].Chemistry A European Journal,2011,17(52):15027-15031.
[8] Xu J,Jung K,Atme A,Shanmugam S,Boyer C.A robust and versatile photoinduced living polymerization of conjugated and unconjugated monomers and its oxygen tolerance[J].Journal of the American Chemical Society,2014,136(14):5508-5519.
[9] Lalevée J,Tehfe M A,Dumur F,Gigmes D,Blanchard N,Morlet-Savary F,Fouassier J P.Iridium photocatalysts in free radical photopolymerization under visible lights [J].ACS Macro Letters,2012,1(2):286-290.
[10] Ke M R,Yeung S L,Fong W P,Ng D K P,Lo P C,A phthalocyanine-peptide conjugate with high in vitro photodynamic activity and enhanced in vivo tumor-retention property[J].Chemistry A European Journal,2012,18(14):4225-4233.
[11] Naik A,Rubbiani R,Gasser G,Spingler B.Visible-lightinduced annihilation of tumor cells with platinum-porphyrin conjugates[J].Angewandte Chemie International Edition,2014,53(27):6938-6941.
[12] Gorman A,Killoran J,O′Shea C,Kenna T,Gallagher W M,O′Shea D F.In vitro demonstration of the heavy-atom effect for photodynamic therapy[J].Journal of the American Chemical Society,2004,126(34):10619-10631.
[13] Lincoln R,Kohler L,Monro S,Yin H,Stephenson M,Zong R,Chouai A,Dorsey C,Hennigar R,Thummel R P.McFarland S A.Exploitation of long-lived 3IL excited states for metal-organic photodynamic therapy:verification in a metastatic melanoma model[J].Journal of the American Chemical Society,2013,135(45):17161-17175.
[14] Singh-Rachford T N,Castellano F N.Photon upconversion based on sensitized triplet-triplet annihilation[J].Coordination Chemistry Reviews,2010,254(21):2560-257.
[15] Zhao J,Ji S,Guo H.Triplet-triplet annihilation based upconversion:from triplet sensitizers and triplet acceptors to upconversion quantum yields[J].RSC Advances,2011,1(6):937-950.
[16] Zhao J,Wu W,Sun J,Guo S.Triplet photosensitizers:from molecular design to applications [J].Chemical Society Reviews,2013,42(12):5323-5351.
[17] Hammarström L,Johansson O.Expanded bite angles in tridentate ligands.Improving the photophysical properties in bistridentate RuII polypyridine complexes[J].Coordination Chemistry Reviews,2010,254(21):2546-2559.
[18] Zhao J,Ji S,Wu W,Wu W,Guo H,Sun J,Sun H,Liu Y,Li Q,Huang L.Transition metal complexes with strong absorption of visible light and long-lived triplet excited states:from molecular design to applications [J].RSC Advances,2012,2(5):1712-1728.
[19] Whittle C E,Weinstein J A,George M W,Schanze K S.Photophysics of diimine platinum(II)bis-acetylide complexes[J].Inorganic Chemistry,2001,40(16):4053-4062.
[20] Hissler M,Connick W B,Geiger D K,McGarrah J E,Lipa D,Lachicotte R J,Eisenberg R.Platinum diimine bis(acetylide)complexes:synthesis,characterization,and luminescence properties[J].Inorganic Chemistry,2000,39(3):447-457.
[21] Pan Y,Kee C W,Chen L,Tan C H.Dehydrogenative coupling reactions catalysed by Rose Bengal using visible light irradiation [J].Green Chemistry,2011,13(10):2682-2685.
[22] Pan Y,Wang S,Kee C W,Dubuisson E,Yang Y,Loh K P,Tan C H.Graphene oxide and Rose Bengal:oxidative C-H functionalisation of tertiary amines using visible light[J].Green Chemistry,2011,13(12):3341-3344.
[23] Ulrich G,Ziessel R,Harriman A.The chemistry of fluorescent bodipy dyes:versatility unsurpassed [J].Angewandte Chemie International Edition,2008,47(7):1184-1201.
[24] Ziessel R,Harriman A.Artificial light-harvesting antennae:electronic energy transfer by way of molecular funnels[J].Chemical Communications,2011,47(2):611-631.
[25] Lu H,Mack J,Yang Y,Shen Z.Structural modification strategies for the rational design of red/NIR region BODIPYs[J].Chemical Society Reviews,2014,43(13):4778-4823.
[26] Loudet A,Burgess K.BODIPY dyes and their derivatives:syntheses and spectroscopic properties [J].Chemical Reviews,2007,107(11):4891-4932.
[27] Yogo T,Urano Y,Ishitsuka Y,Maniwa F,Nagano T.Highly efficient and photostable photosensitizer based on BODIPY chromophore [J].Journal of the American Chemical Society,2005,127(35):12162-12163.
[28] Kamkaew A,Lim S H,Lee H B,Kiew L V,Chung L Y,Burgess K.BODIPY dyes in photodynamic therapy [J].Chemical Society Reviews,2013,42(1):77-88.
[29] Huang L,Yu X,Wu W,Zhao J.Styryl Bodipy-C60dyads as efficient heavy-atom-free organic triplet photosensitizers[J].Organic Letters,2012,14(10):2594-2597.
[30] Ziessel R,Allen B D,Rewinska D B,Harriman A.Selective triplet-state formation during charge recombination in a fullerene/Bodipy molecular dyad (Bodipy= Borondipyrromethene)[J].Chemistry A European Journal,2009,15(30):7382-7393.
[31] Huang L,Cui X,Therrien B,Zhao J.Energy-funnelingbased broadband visible-light-absorbing Bodipy-C60triads and tetrads as dual functional heavy-atom-free organic triplet photosensitizers for photocatalytic organic reactions[J].Chemistry A European Journal,2013,19(51):17472-17482.
[32] Nicewicz D A,MacMillan D W C.Merging photoredox catalysis with organocatalysis:the direct asymmetric alkylation of aldehydes[J].Science,2008,322(5898):77-80.
[33] Yamakoshi Y,Umezawa N,Ryu A,Arakane K,Miyata N,Goda Y,Masumizu T,Nagano T.Active oxygen species generated from photoexcited fullerene(C60)as potential medicines:O2-·versus1O2[J].Journal of the American Chemical Society,2003,125(42):12803-12809.
[34] Gu X,Li X,Chai Y,Yang Q,Li P,Yao Y.A simple metal-free catalytic sulfoxidation under visible light and air[J].Green Chemistry,2013,15(2):357-361.
[35] Zhang P,Wang Y,Li H,Antonietti M.Metal-free oxidation of sulfides by carbon nitride with visible light illumination at room temperature[J].Green Chemistry,2012,14(7):1904-1908.
[36] Dad′ováJ,SvobodováE,Sikorski M,König B,Cibulka R.Photooxidation of sulfides to sulfoxides mediated by tetra-O acetylriboflavin and visible light [J].Chem-CatChem,2012,4(5):620-623.
[37] Baciocchi E,Giacco T D,Elisei F,Gerini M F,Guerra M,Lapi A,Liberali P.Electron transfer and singlet oxygen mechanisms in the photooxygenation of dibutyl sulfide and thioanisole in MeCN sensitized by N-methylquinolinium tetrafluoborate and 9,10-dicyanoanthracene.The probable involvement of a thiadioxirane intermediate in electron transfer photooxygenations[J].Journal of the American Chemical Society,2003,125(52):16444-16454.
[38] Bonesi S M,Manet I,Freccero M,Fagnoni M,Albini A.Photosensitized oxidation of sulfides:discriminating between the singlet oxygen mechanism and electron transfer involving superoxide anion or molecular oxygen[J].Chemistry A European Journal,2006,12(16):4844-4857.
[39] Liu Q,Li Y N,Zhang H H,Chen B,Tung C H,Wu L Z.Reactivity and mechanistic insight into visible-light-induced aerobic cross dehydrogenative coupling reaction by organophotocatalysts[J].Chemistry A European Journal,2012,18(2):620-627.
