单中心174例原发性免疫缺陷病临床预警症状的初步探讨
2013-12-25贺建新江载芳
张 金 贺建新 江载芳 刘 钢
·论著·
单中心174例原发性免疫缺陷病临床预警症状的初步探讨
张 金 贺建新 江载芳 刘 钢
目的 分析并总结原发性免疫缺陷病(PID)患儿的临床感染特征和预警症状,了解预警症状对PID早期识别的应用价值。方法 参考2011年免疫学会国际联合会(IUIS)PID分类委员会公布的方案、泛美免疫缺陷病组(PAGID)和欧洲免疫缺陷病协会(ESID)提出的PID诊断和分类标准,在首都医科大学附属北京儿童医院2000年10月至2011年11月病例检索系统检索出院诊断中含有上述PID分类疾病的病历,对于诊断低丙种球蛋白血症和联合免疫缺陷的患儿除外继发性免疫缺陷病,逐份查阅病历重新诊断,并做出明确、可以和可能诊断,以明确、可以诊断的病例进行预警症状的分析。结果 ①174例PID患儿进入分析,男女比例为4.4∶1,其中抗体缺陷为主的免疫缺陷101例(58.0%),严重联合免疫缺陷病(SCID)34例(19.5%),吞噬细胞功能缺陷19例(10.9%),定义明确的免疫缺陷综合征10例(5.7%),免疫失调性疾病10例(5.7%)。②75例(43.1%)存在反复呼吸道感染,以抗体缺陷为主的免疫缺陷最为常见,与SCID间差异有统计学意义;卡介苗接种后异常反应在慢性肉芽肿病(CGD)中最多见,与抗体缺陷为主的免疫缺陷和SCID比较差异有统计学意义;腹泻病在定义明确的免疫缺陷综合征中较常见,败血症在SCID和CGD患儿中较常见,但PID各类型间比较差异无统计学意义。③72例(41.4%)患儿存在营养发育落后,PID各类型间差异无统计学意义;淋巴结、肝和脾肿大以CGD和免疫失调性疾病最为常见;鹅口疮在SCID中常见,与抗体缺陷为主的免疫缺陷差异有统计学意义;肛周脓肿以CGD多见,与其他PID类型比较差异有统计学意义。107例(61.5%)有明确微生物学证据。④PID患儿共电话随访到85例(48.8%),其中死亡28例(32.9%)。⑤124例为明确和可以诊断PID,其中106例(85.5%)具备≥2条预警症状。静脉应用抗生素清除病灶(96.0%)、体重不增或生长发育极度迟缓(41.1%)、反复呼吸道感染(41.9%)和PID家族史(22.6%)在不同类型PID中均占有较高的比例。结论 预警症状对PID有着很好的提示作用,需要静脉应用抗生素清除病灶、体重不增或生长发育极度迟缓和PID家族史对PID有预警意义,中耳炎、中枢神经系统感染和反复呼吸道感染在抗体缺陷为主的免疫缺陷中较为多见, 深部脓肿、卡介苗接种后异常反应对CGD有预警意义。慢性反复发作性腹泻对PID预警作用值得进一步关注。
原发性免疫缺陷病; 预警症状; 儿童
原发性免疫缺陷病(PID)是指因免疫活性细胞和免疫活性因子先天发育不全引起的免疫反应缺如或降低,导致机体抗感染免疫功能低下的一组临床综合征。免疫缺陷患儿因反复慢性感染极易伴发其他疾病,很多患儿在婴幼儿时期夭折,误诊、漏诊及延误治疗情况突出[1]。为了早期识别PID,Jeffrey Model基金会根据临床研究提出了儿童PID十大临床预警症状[2],但其临床应用价值尚未明确。Subbarayan 等[3]对英国北部的2个儿童免疫缺陷病诊疗中心的PID患儿进行了研究,发现只有3个预警症状对识别PID有帮助。MacGinnitie等[4]研究发现23%诊断为PID的患儿对预警症状不具有敏感性和特异性。目前国内尚无关于PID预警症状的研究,为了更好地了解预警症状的应用价值,本文对首都医科大学附属北京儿童医院(我院)近10余年来收治的PID患儿的临床资料进行回顾性分析,旨在总结临床感染特征及预警症状,以利于早期发现和识别PID,尽早诊断和治疗。
1 方法
1.1 PID诊断和分类标准 参考2011年免疫学会国际联合会(IUIS)PID分类委员会公布的方案、泛美免疫缺陷病组(PAGID)和欧洲免疫缺陷病协会(ESID)1999年提出的PID诊断和分类标准[5,6]。PID分类:①抗体缺陷为主的免疫缺陷:X连锁无丙种球蛋白血症(XLA)、普通变异型免疫缺陷病(CVID)、X连锁高IgM综合征(XHIM)、高IgM综合征(HIGM)、IgA缺乏症(IgAD)、选择性IgM缺乏症(SIgMD)、婴儿期短暂低丙种球蛋白血症(THI)等;②联合免疫缺陷:严重联合免疫缺陷病(SCID)、X连锁严重联合免疫缺陷病(XSCID)、Omenn综合征等;③定义明确的免疫缺陷综合征:湿疹血小板减少伴免疫缺陷综合征(WAS)、共济失调毛细血管扩张综合征(AT)、DiGeorge综合征、高IgE综合征(HIES)等;④吞噬细胞数量和(或)功能缺陷:慢性肉芽肿病(CGD)、白细胞黏附缺陷等;⑤免疫失调性疾病:X连锁淋巴细胞异常增生症(XLP)、家族性噬血细胞性淋巴组织细胞增生症(FLH)、Chediak-Higashi综合征(CHS)等;⑥自身炎症性疾病:Blau综合征等;⑦固有免疫缺陷;⑧补体缺陷。
需要说明的是,由于HIGM临床多以低丙种球蛋白血症发现,故本研究仍将其归为抗体缺陷为主的免疫缺陷。
1.2 病历筛选原则 首先,在我院病例检索系统以上述PID分类的病名逐个在出院诊断中检索。然后,对于检索出的病历中诊断低丙种球蛋白血症和联合免疫缺陷的患儿除外继发性免疫缺陷病,继发因素包括[6],①感染因素:HIV、CMV、EBV感染等;②系统性疾病:免疫球蛋白分解过度导致的免疫缺陷、免疫球蛋白丢失过度导致的免疫缺陷(肾病、严重烧伤、淋巴管扩张、严重腹泻);③药物因素:卡托普利、卡马西平、糖皮质激素、双氯灭酸、苯妥英钠、柳氮磺吡啶等;④恶性肿瘤:慢性淋巴细胞性白血病、非霍奇金淋巴瘤、B细胞性恶性肿瘤等;⑤染色体异常:18q-综合征、22单体综合征、18三体综合征、唐氏综合征。最后,对筛选出的病历,逐份查阅病历依据文献[5,6]重新诊断,并做出明确诊断、可以诊断和可能诊断,对不符合PID诊断的病例予以排除。
1.3 临床数据采集与定义
1.3.1 一般情况 性别,发病年龄,诊断年龄,诊断时间。
1.3.2 家族史 查阅病史记录的家族史行一、二、三级亲属分类。

1.3.4 合并症 是指PID除感染以外的疾病[10,11],包括肺部疾病、自身免疫性疾病、血液系统疾病和肿瘤等。
1.3.5 预警症状 Jeffrey Model基金会提出的儿童PID十大临床预警症状[2]:①1年内中耳感染次数>4次;②1年内严重鼻窦感染>2次;③抗生素治疗2个月疗效不佳;④1年内患肺炎>2次;⑤婴幼儿体重不增或生长发育极度迟缓;⑥反复深部皮肤或器官脓肿;⑦持续鹅口疮或皮肤真菌感染;⑧需要静脉应用抗生素以清除感染灶;⑨≥2处的顽固性感染(包括败血症);⑩有PID家族史。本研究结合中国儿童感染实际情况,将②和④条合并至①中,表示为反复呼吸道感染,故本文儿童PID预警症状为8条。
1.3.6 病原学 来自于病史记录中的血、痰、肺泡灌洗液、胸腔积液、脑脊液、骨髓等培养结果。
1.4 转归 对进入分析的PID病历进行电话访问,了解患儿疾病转归情况。
1.5 统计学方法 采用SPSS 17.0软件进行统计学数据处理。各疾病类型临床特点的比较采用行×列χ2检验或Fisher精确概率法,P<0.05为差异具有统计学意义,进一步分析两组间的差异采用χ2分割法,P<α为差异具有统计学意义,α=0.05/[n(n-1)/2],n为分割后组数。
2 结果
2.1 PID病历筛选 2000年10月至2011年11月我院出院诊断中含有上述PID分类疾病的病历900例,排除继发性免疫缺陷病411例,依据文献[5,6]不符合可能诊断296例,因THI(14例)临床症状较轻,Blau综合征(5例)感染特征不明显、表现复杂,固有免疫和补体缺陷未检出,本研究病例筛选见图1。
2.2 PID重新诊断结果 文献[5,6]8个PID分类标准中,表1显示,本文174例PID涉及其中的5类15个PID病种(具体诊断标准见文后附件)。174例PID中明确诊断33例,可以诊断91例,可能诊断50例。需要说明的是,SIgMD、HIES和CHS在文献[5,6]中没有明确诊断、可以诊断和可能诊断的区分。SIgMD符合相应诊断纳入明确诊断中;HIES根据临床评分标准,>40分为临床诊断,纳入可以诊断中;CHS符合中性粒细胞及其前体细胞内特征性异常的粗大溶酶体颗粒,纳入明确诊断中。

图1 病例筛选流程图
Fig 1 Flow chart of study subjects
表1 174例PID患儿类型及家族史情况[n(%)]
Tab 1 The classification and family history of 174 PID patients[n(%)]
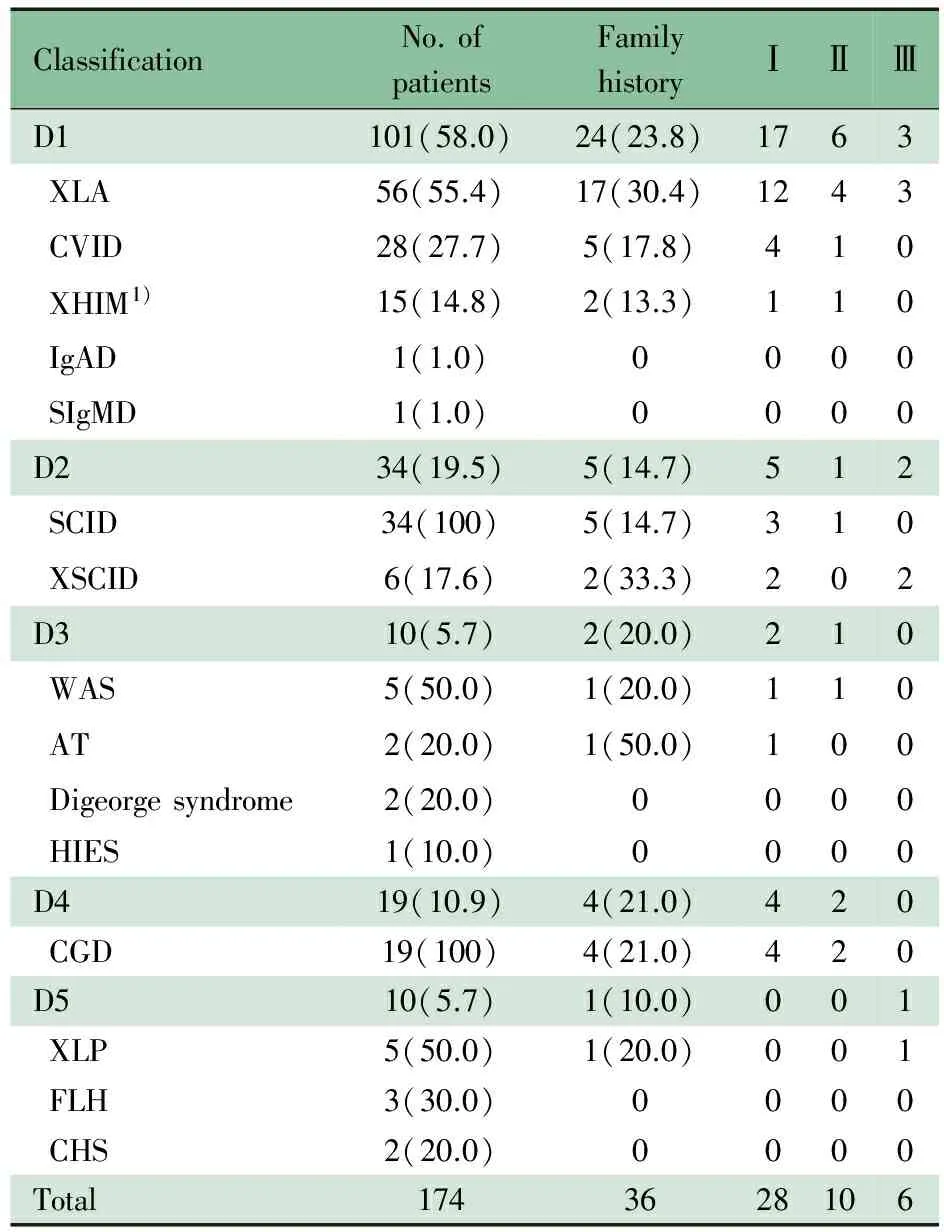
ClassificationNo.ofpatientsFamilyhistoryⅠⅡⅢD1101(58.0)24(23.8)1763XLA56(55.4)17(30.4)1243CVID28(27.7)5(17.8)410XHIM1)15(14.8)2(13.3)110IgAD1(1.0)0000SIgMD1(1.0)0000D234(19.5)5(14.7)512SCID34(100)5(14.7)310XSCID6(17.6)2(33.3)202D310(5.7)2(20.0)210WAS5(50.0)1(20.0)110AT2(20.0)1(50.0)100Digeorgesyndrome2(20.0)0000HIES1(10.0)0000D419(10.9)4(21.0)420CGD19(100)4(21.0)420D510(5.7)1(10.0)001XLP5(50.0)1(20.0)001FLH3(30.0)0000CHS2(20.0)0000Total1743628106
Notes D1: predominant antibody deficiencies; D2: combined T and B cell immunodeficiencies; D3: other well-defined immunodeficiency syndromes; D4: congenital defects of phagocyte; D5: diseases of immune dysregulaton. Ⅰ: first degree relatives, such as parents and siblings; Ⅱ : second degree relatives, such as grandparents and uncles; Ⅲ: third degree relatives,such as cousins. 1) one case of XHIM was a girl,who was diagnosed as HIGM
2.3 PID类型与家族史 表1显示,36例(20.7%)PID病例存在阳性家族史,其中一级亲属28例(77.8%),二级亲属10例(27.8%),三级亲属6例(16.7%);不同分类PID阳性家族史构成比差异无统计学意义 (P=0.568)。
表2 各疾病类型发病及诊断年龄中位数及范围(月)
Tab 2 The age of onset and diagnosis of PID patients (months)
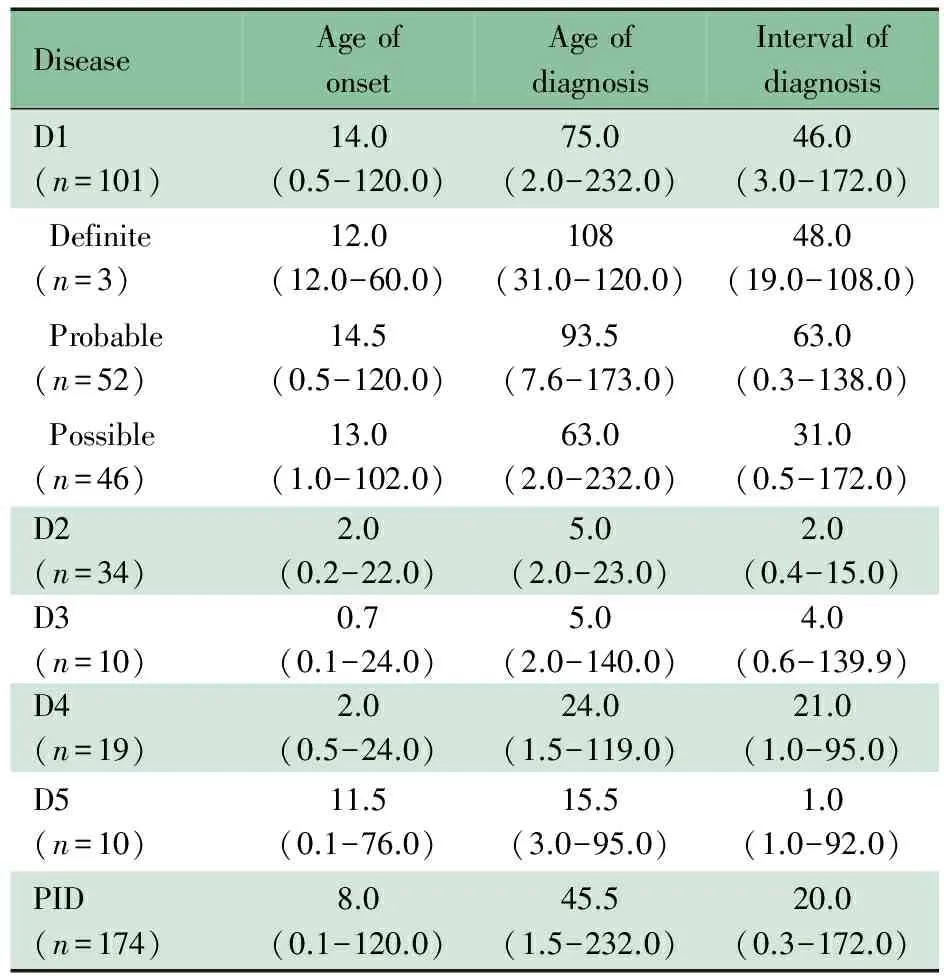
DiseaseAgeofonsetAgeofdiagnosisIntervalofdiagnosisD1(n=101)14.0(0.5-120.0)75.0(2.0-232.0)46.0(3.0-172.0)Definite(n=3)12.0(12.0-60.0)108(31.0-120.0)48.0(19.0-108.0)Probable(n=52)14.5(0.5-120.0)93.5(7.6-173.0)63.0(0.3-138.0)Possible(n=46)13.0(1.0-102.0)63.0(2.0-232.0)31.0(0.5-172.0)D2(n=34)2.0(0.2-22.0)5.0(2.0-23.0)2.0(0.4-15.0)D3(n=10)0.7(0.1-24.0)5.0(2.0-140.0)4.0(0.6-139.9)D4(n=19)2.0(0.5-24.0)24.0(1.5-119.0)21.0(1.0-95.0)D5(n=10)11.5(0.1-76.0)15.5(3.0-95.0)1.0(1.0-92.0)PID(n=174)8.0(0.1-120.0)45.5(1.5-232.0)20.0(0.3-172.0)
Notes D1: predominant antibody deficiencies; D2: combined T and B cell immunodeficiencies; D3: other well-defined immunodeficiency syndromes; D4: congenital defects of phagocyte; D5: diseases of immune dysregulaton
2.4 PID类型与发病和诊断年龄 表2显示,本组PID病例总发病年龄的中位数为8个月,最早为生后起病,最大发病年龄为10岁,其中抗体缺陷为主的免疫缺陷和免疫失调性疾病发病年龄相对偏大,发病年龄中位数为12个月左右;联合免疫缺陷、定义明确的免疫缺陷综合征和吞噬细胞功能缺陷多于生后2个月内起病。总的诊断时间中位数为20个月,最长172个月(14.3年),其中以抗体缺陷为主的免疫缺陷诊断时间最长(46个月),其次为吞噬细胞缺陷(21个月)。以抗体缺陷为主的免疫缺陷在明确诊断、可以诊断和可能诊断中发病年龄相似,但明确和可以诊断的患儿诊断时间明显延迟于可能诊断;联合免疫缺陷均为可以诊断病例;吞噬细胞功能缺陷均为明确诊断病例;定义明确的免疫缺陷综合征中明确诊断3例,可以诊断4例,可能诊断3例;免疫失调性疾病明确诊断9例,可能诊断1例。
2.5 PID感染疾病特征 表3显示,75例(43.1%)患儿存在反复呼吸道感染,以抗体缺陷为主的免疫缺陷最为常见(60.4%),与SCID间差异有统计学意义(P=0.000),与其他PID类型差异无统计学意义;中耳炎在抗体缺陷为主的免疫缺陷中比例最高(25.7%),其次为CGD(10.6%),两类型间差异无统计学意义。中枢神经系统感染以抗体缺陷为主的免疫缺陷最为常见(26.7%)与SCID差异有统计学意义(P=0.003);卡介苗接种后异常反应在CGD中最常见(47.4%),与抗体缺陷为主的免疫缺陷和SCID比较差异有统计学意义(P分别为0.000和0.001),定义明确的免疫缺陷综合征和免疫失调性疾病例数少,未行统计学分析。腹泻病在定义明确的免疫缺陷综合征中比例较高(50.0%),败血症在SCID和CGD患儿中较常见(26.3%和17.3%),但PID各类型间比较差异无统计学意义。皮肤感染相对少见(9.8%)。对抗体缺陷为主的免疫缺陷患儿进一步分析,发现明确和可以诊断患儿与可能诊断患儿相比,反复呼吸道感染较常见(P=0.018),皮肤感染较少见(P<0.001),其他感染性疾病明确和可以诊断与可能诊断间差异无统计学意义。
另外,有3例中耳炎患儿(XLA)感染次数超过2次,1例中枢神经系统感染患儿(XLA)先后发生1次病毒性脑炎和3次化脓性脑膜炎,最后放弃治疗。

表3 各类型常见的感染疾病[n(%)]
Notes D1: predominant antibody deficiencies; D2: combined T and B cell immunodeficiencies; D3: other well-defined immunodeficiency syndromes; D4: congenital defects of phagocyte; D5: diseases of immune dysregulaton. RRTI:recurrent respiratory tract infection; CNSI: central nervous system infection.1) partition of chi-square was used to get a result thatP<α, α=0.05/[n(n-1)/2],n, the number after partition
2.6 PID主要体征 表4显示,72例(41.4%)患儿存在营养发育落后,PID各类型间差异无统计学意义;淋巴结、肝、脾肿大以CGD(78.9%、78.9%、73.7%)和免疫失调性疾病(60.0%、90.0%、80.0%)最为常见,SCID中肝、脾肿大比例也较高(73.5%和50.0%);鹅口疮在SCID中多见(32.4%),与抗体缺陷为主的免疫缺陷差异有统计学意义(P=0.002);皮疹在定义明确的免疫缺陷综合征中最为常见(60.0%),与CGD比较差异有统计学意义(P=0.003),与其他PID类型差异无统计学意义;肛周脓肿仅在CGD和抗体缺陷为主的免疫缺陷中发现,以CGD多见(47.4%),与其他PID类型比较差异均有统计学意义。在抗体缺陷为主的免疫缺陷患儿中,明确和可以诊断患儿鹅口疮较可能诊断患儿少见,分别为1例(1.8%)和8例(17.4%),其他主要体征在明确和可以诊断与可能诊断间差异无统计学意义。
2.7 PID合并症 101例抗体缺陷为主的免疫缺陷患儿中,免疫相关性关节炎[12]28例(27.7%),贫血24例(23.8%),粒细胞减少23例(22.8%),支气管扩张16例(15.8%),溶血性贫血3例,噬血细胞综合征、骨髓增生异常综合征和淋巴瘤各2例,特发性血小板减少性紫癜(ITP)、皮肤型红斑狼疮、川崎病、炎症性肠病各1例,合并肉芽肿病变1例;34例联合免疫缺陷中,贫血15例(44.1%);10例定义明确的免疫缺陷综合征中,支气管扩张1例,贫血2例,粒细胞减少1例;19例吞噬细胞缺陷中,贫血9例(47.4%),粒细胞减少2例;10例免疫失调性疾病中,贫血8例(80.0%),粒细胞减少8例(80.0%),噬血细胞综合征3例(30.0%)。

表4 各疾病类型常见体征[n(%)]
Notes D1: predominant antibody deficiencies; D2: combined T and B cell immunodeficiencies; D3: other well-defined immunodeficiency syndromes; D4: congenital defects of phagocyte; D5: diseases of immune dysregulaton.1) partition of chi-square was used to get a result thatP<α, α=0.05/[n(n-1)/2],n, the number after partition
2.8 PID预警症状 本研究中符合明确和可以诊断的PID患儿共124例,对其进行预警症状分析。
2.8.1 PID各类型所包含的预警症状 如表5所示,52例(41.9%)具备第1条预警症状,在抗体缺陷为主的免疫缺陷中最为多见(70.9%),与SCID和CGD比较差异有统计学意义(P分别为0.000和0.003);35例(28.2%)具备第6条预警症状,以CGD多见(68.4%),与抗体缺陷为主的免疫缺陷和SCID比较差异有统计学意义(P分别为0.000和0.009);13例(10.5%)具备第7条预警症状,以SCID常见(23.5%),与抗体缺陷为主的免疫缺陷比较差异有统计学意义(P=0.006),与CGD比较无显著差异。22例(17.7%)具备第3条预警症状,其中以免疫失调性疾病最为明显(44.4%);51例(44.1%)具备第5条预警症状,119例(96.0%)具备第8条预警症状,37例(29.8%)具备第9条预警症状、28例(22.6%)有第10条预警症状,第3,5,8~10条预警症状与其他PID类型间差异无统计学意义。
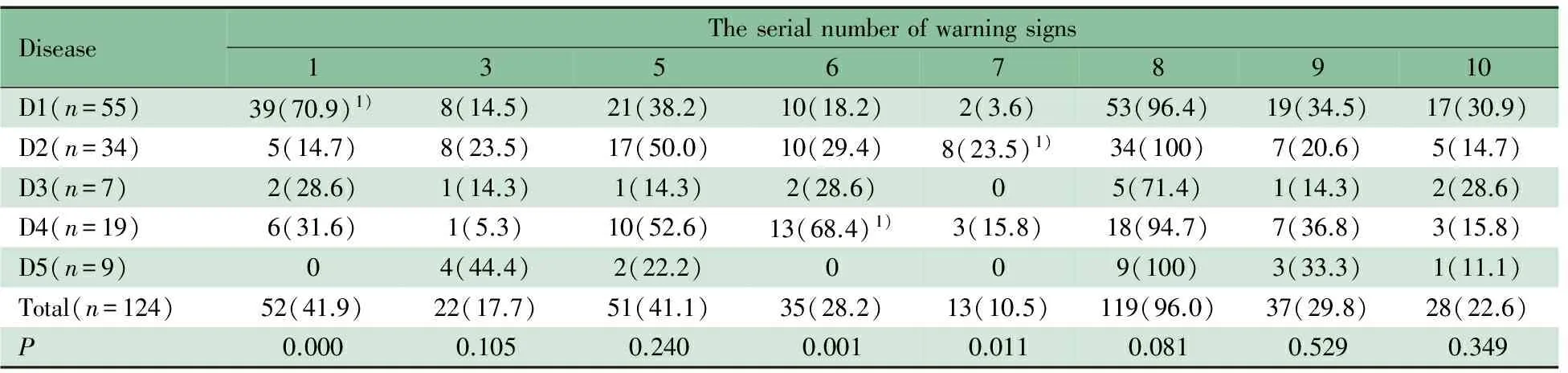
表5 各疾病类型包含预警症状情况[n(%)]
Notes D1: predominant antibody deficiencies; D2: combined T and B cell immunodeficiencies; D3: other well-defined immunodeficiency syndromes; D4: congenital defects of phagocyte; D5: diseases of immune dysregulaton.1,:recurrent respiratory tract infection; 3: ≥2 months on antibiotics with little effect; 5: failure of an infant to gain weight or grow normally; 6: recurrent, deep skin or organ abscesses; 7: persistent thrush in mouth or fungal infection on skin; 8: need for intravenous antibiotics to clear infections; 9:≥2 deep-seated infections including septicemia; 10: family history of primary immunodeficiency.1) partition of chi-square was used to get a result thatP<α, α=0.05/[n(n-1)/2],n, the number after partition
2.8.2 PID各类型所包含的预警症状数目 表6所示,包含最多的预警症状数目为6条,有4例患儿;具备2条及以上的PID患儿106例(85.5%),74例(59.7%)集中在2条和3条。定义明确的免疫缺陷综合征和免疫失调性疾病包含的预警症状较少,最多具备3条或4条。
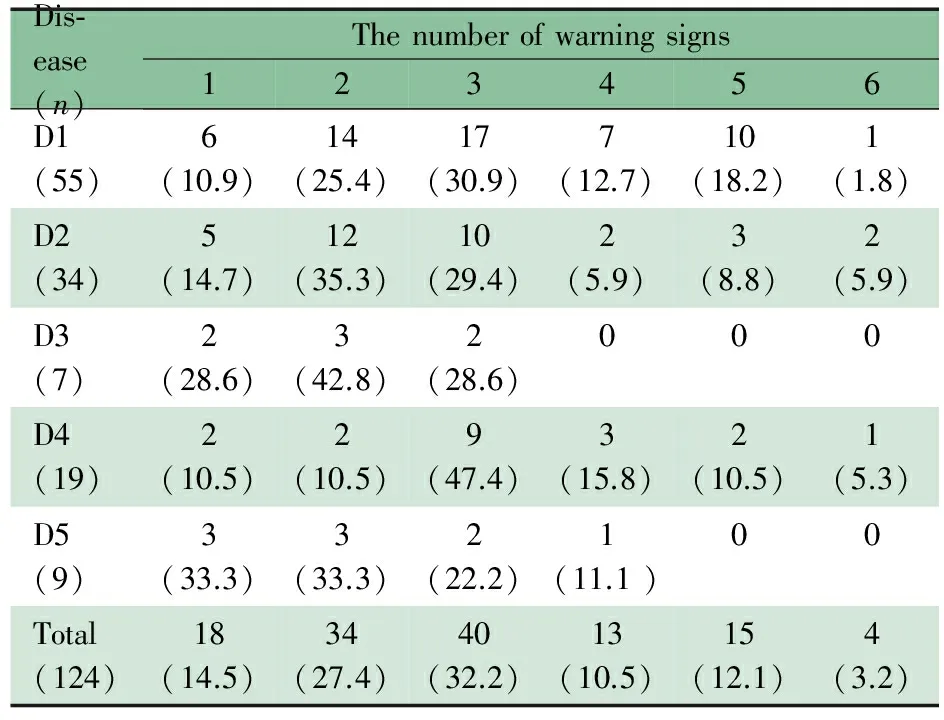
表6 各疾病类型预警症状数目情况[n(%)]
Notes D1: predominantly antibody deficiencies; D2: combined T and B cell immunodeficiencies; D3: other well-defined immunodeficiency syndromes; D4: congenital defects of phagocyte; D5: diseases of immune dysregulaton.
2.9 PID病原学特点 107例(61.5%)有明确微生物学证据,如抗体缺陷为主的免疫缺陷以细菌感染多见,依次为肺炎链球菌18例(17.8%)、铜绿假单胞菌11例(10.9%)和流感嗜血杆菌2例(2.0%)。免疫失调性疾病9例(90.0%)存在病毒感染,分别为EBV 7例,CMV 2例,HHV 6和HHV 7各2例,HSV1例。
2.10 PID转归 85例(48.9%)PID患儿完成了电话随访,其中死亡28例(27.7%),分别为XLA 8例,CVID 1例,HIGM 3例,SCID 7例,CGD 6例和XLP 3例。26例死于严重感染引起的脏器功能衰竭,1例XLA死于骨髓移植失败,1例CVID死于淋巴瘤。3例行骨髓移植,其中1例XLP患儿一般情况尚可,1例HIGM和FLH出现了移植物抗宿主反应。余54例PID患儿予IVIG、抗感染及对症治疗仍存活;13例抗体缺陷为主的免疫缺陷患儿规律使用IVIG,其中8例感染次数较前减少或感染症状减轻,31例未规律使用或未使用IVIG,仍存在反复感染,总体生活质量较差。
3 讨论
近年来,随着医疗水平和诊断技术的进步,国内外关于PID的报道日益增多[13~15]。2005年美国免疫缺陷基金会(IDF)估计人群PID的发病率为1∶1 200,儿童的发病率为1∶2 000[16]。2009年Joshi等[17]首次报道了以人群为基础的PID流行病学结果,估计人群发病率为4.6/10 000。目前国内尚未建立完善的PID登记制度,全国确诊的PID病例少之又少[18],在有关PID患者的研究中,多以出院诊断含有PID的患者作为研究对象,疾病诊断受不同医生临床经验的影响,具有一定的主观性。PAGID和ESID于1999年提出的PID诊断标准简明、客观和明确,以确保不同的医生、科学家以及患者在研究工作中应用相同定义。PAGID和ESID的PID的诊断标准分为:明确、可以和可能诊断标准。有研究表明,符合明确诊断标准PID患者有98%的概率在20年随访后仍维持原有诊断不变。基因检测是获得明确诊断最可信的方法,有些病种缺乏特异的mRNA及其蛋白质产物也有助于其诊断。符合可以诊断标准PID患者仅有临床和实验室表现而无明确的基因异常,也无其相应mRNA或蛋白质缺乏的证据,有85%的概率在20年随访后仍维持原有诊断不变。符合可能诊断标准的患者仅有部分临床和实验室表现。本文以此标准对所有出院诊断含有PID病例进行重新诊断,尽可能减少了PID诊断性偏倚的发生。
本研究PID病例普遍存在诊断延误情况,诊断时间中位数20个月,最长为172个月(14.3)年,比国外报道的1996年后的相关研究平均诊断时间2.7年短[17],可能与本文纳入的是我院晚近10年的PID病例有关。本文抗体缺陷为主的免疫缺陷患儿诊断延误情况最为明显,其次为吞噬细胞功能缺陷。值得思考的是在抗体缺陷为主的免疫缺陷患儿中,对明确、可以和可能诊断的发病及诊断时间分析表明,三者的发病年龄相似,但明确和可以诊断患儿的诊断时间明显延迟于可能诊断,提示目前抗体缺陷为主的免疫缺陷从怀疑到确诊还需很长时间,应该重视可能诊断病例。
随着PID种类的增多及分子生物学的发展,Arkwright等[19]认为10个临床预警症状[2]对于PID的警示作用正在减弱,且未包括一次性感染、自身免疫和炎症性疾病、肿瘤等。MacGinnitie等[4]研究发现23%诊断为PID的患儿对预警症状[2]不具有敏感性和特异性,主要为免疫性和过敏性疾病。本文在预警症状研究中只分析符合明确和可以诊断标准的PID患儿,提高了预警症状的准确度和可信度。结果表明,需静脉应用抗生素清除病灶(96.0%)、体重不增或生长发育极度迟缓(41.1%)、反复呼吸道感染(41.9%)和PID家族史(22.6%)在不同类型PID中都占有较高的比例。这与Subbarayan 等[3]对英国北部的2个儿童免疫缺陷病诊疗中心的PID患儿有关预警症状的评价结果一致,Subbarayan发现仅有需静脉应用抗生素清除病灶、体重不增或生长发育极度迟缓和PID家族史等3个预警症状对识别PID有帮助。需要说明的是,本文出于对国内儿童感染现状的考虑,将1年内中耳感染次数超过4次、1年内严重鼻窦感染超过2次和1年内患肺炎超过2次合并统计为反复呼吸道感染进行分析,国外文献关于反复呼吸道感染的研究不多,其概念、临床特点和诊断标准与我国也不一致。有作者认为6岁以内的儿童1年内通常会有6~12次病毒感染,表现为上呼吸道感染或腹泻,症状轻微且有自限性。在幼儿园或日托中心的儿童更易发生频繁的病毒感染。中耳炎在婴幼儿中较常见,与免疫是否异常无关。儿童期有频繁或反复的轻微疾病(如病毒性感染所致的上呼吸道感染),但不影响生长发育,则不会有潜在的免疫紊乱,导致频繁感染。我国1987年中华医学会儿科学分会呼吸学组制定了《反复呼吸道感染诊断参考标准》,并于2007年对反复呼吸道感染的临床概念和判断条件进行了讨论和修订,强调了反复上、下呼吸道感染的部位,特别是反复气管支气管炎和反复肺炎[20]。反复呼吸道感染大多伴有基础疾病,先天性支气管肺发育异常、遗传因素、PID、感染、环境、护理因素等都是造成反复呼吸道感染的原因。然而,青少年或成人1年2次以上的中耳炎,每年患细菌性肺炎或严重的鼻窦炎,或病史中有2次以上危及生命的重症感染者,提示存在潜在免疫缺陷的可能。由于本文为病例回顾性研究,所有明确和可以诊断PID病例中,中耳炎患儿只有3例感染次数超过2次,其余均为单次记录,病例中对鼻窦炎和肺炎次数的资料不够完整,或病史中仅描写了反复呼吸道感染或呼吸道感染,无法区分上、下呼吸道感染,所以对中耳炎、鼻窦炎和肺炎预警项目进行了合并。有国外报道上呼吸道感染以中耳炎、鼻窦炎常见[17,21],国内则以鼻咽部感染为主,中耳炎、鼻窦炎少见[22],而下呼吸道感染差异不大,本文发现反复呼吸道感染、中耳炎和中枢神经系统感染在抗体缺陷为主的免疫缺陷中较为多见,可出现身体多个无菌部位侵袭性感染,反复呼吸道感染应是临床不容忽视的预警症状。
本研究发现,各类PID中有高达22.4%患儿出现慢性腹泻,定义明确的免疫缺陷综合征病例50%均合并慢性腹泻,有研究显示慢性腹泻在PID患者中的发生率明显高于非PID患者[7]。国外一项研究中27% PID患者存在胃肠道功能紊乱,且多数为复发性胃肠道感染[17],有报道显示与IVIG存在相关性。慢性腹泻能否作为PID相关预警症状,有待收集更多资料进一步分析。
本文中接种减毒活疫苗后出现异常反应的患儿占8.0%,尤其在CGD患儿中十分常见(47.4%),本文CGD患儿中出现的均为卡介苗接种后异常反应,文献报道卡介苗接种后异常反应病例中有半数患有PID[23,24],多见于SCID、CGD和某些特定的免疫缺陷综合征等。我国新生儿出生后常规接种卡介苗,接种后出现异常反应,尤其是播散性卡介苗病应引起高度重视,应重视卡介苗接种后异常反应对CGD的预警意义。需要注意的是22.6%病例均有PID家族史,且多为一级亲属,CGD病例中有3例(15.8%)也有家族史,本文无法得到家族史中有关减毒活疫苗接种后出现异常反应的病史,至少应对于有类似家族史的患儿谨慎接种卡介苗。
本文抗体缺陷为主的免疫缺陷患儿中免疫相关性关节炎发生率较高,其次为溶血性贫血、ITP、红斑狼疮。本文XLA患儿中发现1例川崎病,XLA合并川崎病较少见,仅有个别文献报道[25]。2例淋巴瘤患儿均为CVID,其中1例明确诊断为霍奇金淋巴瘤,与文献报道的CVID患儿肿瘤发生率升高相符,但文献以非霍奇金淋巴瘤多见[26]。故对伴发或只表现为自身免疫性疾病、肿瘤的患儿,亦应警惕免PID的可能。
本研究中,10例免疫失调性疾病患儿中有9例存在病毒感染, 4例(44.4%)抗生素治疗2个月疗效不佳,值得临床医生重视,这些患儿多具备肝、脾和淋巴结肿大,临床很难除外败血症,但也应重视免疫失调性疾病的可能,这些免疫失调性疾病患儿同胞中会出现致死性病毒感染的家族史,应重视阳性家族史病例,尽早进行家族筛查,早期发现并指导优生优育。
Jeffrey Model基金会提出的儿童PID十大临床预警症状,≥2条提示临床医师应警惕PID的发生。本研究对PID各类型所包含的预警症状进行了统计,发现≥2条的患儿比例为85.5%,说明预警症状对PID具有很好的警示作用,本研究中59.7%的PID病例集中在2条或3条,因此当出现2条或3条,甚至1条预警症状时就应该警惕PID。
本文预警症状结果分析表明,需要静脉应用抗生素清除病灶、体重不增或生长发育极度迟缓和PID家族史对PID有预警意义,中耳炎、中枢神经系统感染和反复呼吸道感染在抗体缺陷为主的免疫缺陷中较为多见,深部脓肿、卡介苗接种后异常反应对CGD有预警意义。慢性反复发作性腹泻对PID预警作用值得进一步关注,对伴发或只表现为自身免疫性疾病、肿瘤的患儿,亦应警惕PID。临床医生应提高对预警信号的重视程度,争取早期识别PID,有条件应进行基因诊断。
本研究的不足与局限,①本文为单中心病例,虽然收集了近10年病例,PID样本量仍然很小,病种亦不全面。②本文中临床资料来源于住院PID患儿,在门诊诊断和治疗的PID患儿数据无法采集。③本文为回顾性研究,PID患儿的结局仅进行了一次电话预后随访。④患儿的临床症状和家族史资料来自于病历描述,但实际可能存在更多的阳性表现。
[1]Eades-Perner AM, Gathmann B, Knerr V, et al. The European internet-based patient and research database for primary immunodeficiencies: results 2004-06. Clin Exp Immunol,2007,147(2):306-312
[2]Cooper MA, Pommering TL, Koranyi K. Primary immunodeficiencies. Am Fam Physician, 2003, 68(10): 2001-2008
[3]Subbarayan A, Colarusso G, Hughes SM, et al. Clinical features that identify children with primary immunodeficiency diseases. Pediatrics,2011, 127(5): 810-816
[4]MacGinnitie A, Aloi F, Mishra S. Clinical characteristics of pediatric patients evaluated for primary immunodeficiency. Pediatr Allergy Immunol,2011, 22(7): 671-675
[5]Al-Herz W, Bousfiha A, Casanova JL, et al. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Front Immunol,2011, 2:54
[6]Conley ME, Notarangelo LD, Etzioni A. Diagnostic criteria for primary immunodeficiencies. Representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies). Clin Immunol, 1999, 93(3): 190-197
[7]Yarmohammadi H, Estrella L, Doucette J, et al. Recognizing primary immune deficiency in clinical practice. Clin Vaccine Immunol, 2006, 13(3): 329-332
[8] Zhao HJ(赵惠君 ), Chen TX, Hao YQ, et al. Overview of clinical occurrence of primary immunodeficiency disorders in children. Chin J Pediatr(中华儿科杂志), 2006, 44(6): 403-406
[9]Li H(李辉), Ji CY, Zong XN, et al.Height and weight standardized growth charts for Chinese children and adolescents aged 0 to 18 years.Chin J Pediatr(中华儿科杂志), 2009,47(7):487-492
[10]Cao LZ(曹励之), Wang Z.Primary immunodeficiency diseases of hematologic manifestation.J Appl Clin Pediatr(实用儿科临床杂志),2008,23(3):169 -172
[11]Goyal R, Bulua AC, Nikolov NP, et al. Rheumatologic and autoimmune manifestations of primary immunodeficiency disorders. Curr Opin Rheumatol, 2009,21(1):78-84
[12]Verbruggen G, De Backer S, Deforce D, et al. X linked agammaglobulinaemia and rheumatoid arthritis. Ann Rheum Dis,2005, 64(7): 1075-1078
[13] Shabestari M S, Maljaei S H, Baradaran R, et a1. Distribution of primary immunodeficiency diseases in the Turk Ethnic Group.living in the Northwestern Iran. J Clin Immunol,2007,27(5): 510-516
[14]Kirkpatrick P, Riminton S. Primary immunodeficiency diseases in Australia and New Zealand. J Clin Immunol, 2007, 27(5): 517-524
[15]Leiva LE, Zelazco M, Oleastro M, et al. Primary immunodeficiency diseases in Latin America: the second report of the LAGID registry. J Clin Immunol, 2007, 27(1): 101-108
[16]Boyle JM, Buckley RH. Population prevalence of diagnosed primary immunodeficiency diseases in the United States. J Clin lmmunol,2007, 27(5): 497-502
[17]Joshi AY, Iyer VN, Hagan JB,et al.Incidence and temporal trends of primary immunodeficiency: a population-based cohort study. Mayo Clin Proc,2009, 84(1): 16-22
[18]Chen TX(陈同辛).Early recognition and intervention of primary immunodeficiency.Chin J Pediatr(中华儿科杂志),2006,44(6):427-430
[19]Arkwright PD, Gennery AR. Ten warning signs of primary immunodeficiency: a new paradigm is needed for the 21st century. Ann N Y Acad Sci,2011, 1238: 7-14
[20]Chen HZ(陈慧中), Hu YJ.How should we understand the recurrent respiratory tract infections?.Chin J Pediatr(中华儿科杂志),2008,46(2):83-84
[21]Conley ME, Howard V. Clinical findings leading to the diagnosis of X-linked agammaglobulinemia. J Pediatr, 2002, 141(4):566-571
[22]Wang XC(王晓川).Clinical features of X-linked agammaglobulinemia:analysis of 8 cases.Chin J Pediatr(中华儿科杂志),2004,42(8):564-567
[23]He JX(贺建新),Zhao SY,Jiang ZF, et al. Severe Bacillus Caunette-Guerin lymphadenitis and X-linked chronic granulomatous disease in children.Chin J Contemp Pediatr(中国当代儿科杂志),2010,12(6):490-493
[24]Shen X(沈鑫),Zhang JG, Mei J, et al.全身播散性卡介苗感染. Sh J Prev Med(上海预防医学),2002,14(6):299-300
[25]Behniafard N, Aghamohammadi A, Abolhassani H, et al. Autoimmunity in X-linked agammaglobulinemia: Kawasaki disease and review of the literature. Expert Rev Clin Immunol,2012, 8(2): 155-159
[26]Deane S, Selmi C, Naguwa SM,et al. Common variable immunodeficiency: etiological and treatment issues. Int Arch Allergy Immunol, 2009;,150(4):311-324
Preliminary study of the warning signs of 174 children with primary immunodeficiency diseases from a single center
ZHANGJin,HEJian-xin,JIANGZai-fang,LIUGang
(BeijingChildren'sHospitalofCapitalMedicalUniversity,Beijing100045,China)
LIU Gang,E-mail:liugang10@hotmail.com
ObjectiveTo analyze and summarize the clinical features and the warning signs of children with PID, in order to investigate the application value of the warning signs to early identification of PID.Methods According to the classification issued by the International Union of Immunological Societies (IUIS) Primary Immunodeficiency Diseases (PID) Classification Committee in 2011 and the diagnostic criteria made by Pan-American Group for Immunodeficiency (PAGID) and European Society for Immunodeficiencies (ESID), the cases diagnosed as PID above were retrieved . Secondary immunodeficiencies about the cases of hypogammaglobulinemia and combined T and B cell immunodeficiency were excluded. All cases screened out were read, definite, probable or possible diagnoses were made. At last, the warning signs of cases with both definite and probable diagnosis were analyzed.Results 174 children with PID were included, the boys/girls ratio was 4.4:1. Predominantly antibody deficiencies were the most frequent finding (58.0%), followed by combined T and B cell immunedeficiencies (19.5%), congenital defects of phagocyte (10.9%), other well-defined immunodeficiency syndromes (5.7%) and diseases of immune dysregulaton (5.7%). 75 children (43.1%) had a history of recurrent respiratory tract infections, which were common in predominant antibody deficiencies. Diarrheal diseases were common in other well-defined immunodeficiency syndromes. Sepsis was more common in children with SCID and CGD. The occurrence of abnormal reactions after BCG vaccination was particularly high in CGD and significantly different from other types. Malnutrition was very common in 72 children (41.4%) without significant differences. There were significant differences in signs of lymphadenectasis and hepatosplenomegaly in which CGD and diseases of immune dysregulaton were the most common. Thrush was common in SCID. Nine children (47.4%) with CGD were found presenting perianal abscess, which was more common than other types. Pathogen evidence was found in 107 children(61.5%). 85 cases (48.8%) were followed up and 28(32.9%) were dead. There were 124 cases meeting the definite or probable diagnosis, among them 106(85.5%) had 2 or more warning signs. 119 children (96.0%) needed intravenous antibiotics to clear infections. 51 children (41.1%) failed to gain weight or grow normally. 52 children (41.9%) were suffering from recurrent respiratory tract infections. 28 children(22.6%) had a family history of PID.ConclusionThe warning signs can be good indictors to PID. Need for intravenous antibiotics, failure to thrive and family history are useful to early identification of PID. Otitis media, central nervous system infection, sepsis and recurrent respiratory tract infection are more common in predominant antibody deficiencies. Deep-seated infections and abnormal reaction after BCG vaccination are significant to CGD. High attention is expected to be paid to chronic recurrent diarrhea.
Primary immunodeficiency disease; Warning signs; Children
首都医科大学附属北京儿童医院 北京,100045
刘钢,E-mail: liugang10@hotmail.com
10.3969/j.issn.1673-5501.2013.06.007
附录:15个PID的诊断标准
1 X连锁无丙种球蛋白血症(XLA)
1.1 明确诊断 男性,CD19+B细胞<0.02,并符合以下至少1项:①Btk基因突变;②Northern blot检测中性粒细胞或单核细胞发现缺乏Btk mRNA;③单核细胞或血小板缺乏Btk蛋白;④母系的表兄、舅舅或外甥CD19+B细胞<0.02。
1.2 可以诊断 男性,CD19+B细胞<0.02,并符合以下全部标准:①生后5年内表现为反复细菌感染;②血清IgG、IgM和IgA水平≤相应年龄正常值2s;③缺乏同族血凝素和(或)对疫苗应答反应差;④排除其他可导致低丙种球蛋白血症的原因。
1.3 可能诊断 男性,CD19+B细胞<0.02,排除其他可导致低丙种球蛋白血症的原因,并符合以下至少1项:①生后5年内表现为反复细菌感染;②血清IgG、IgM 和IgA水平≤相应年龄正常值2s;③缺乏同族血凝素。
2 普通变异型免疫缺陷(CVID)
2.1 可以诊断 血清IgG水平明显降低(≤相应年龄均值2s),且IgA和(或)IgM水平明显降低,并符合以下全部条件:①2岁以后发病;②缺乏同族血凝素和(或)对疫苗应答反应差;③排除其他可导致低丙种球蛋白血症的原因。
2.2 可能诊断 血清同种型免疫球蛋白(IgM、IgG、IgA)中一种明显降低(≤相应年龄均值2s),并符合以下全部条件:①2岁以后发病;②缺乏同族血凝素和(或)对疫苗应答反应差;③排除其他可导致低丙种球蛋白血症病因。
3 X连锁高IgM综合征(XHIM)
3.1 明确诊断 男性,血清IgG水平≤相应年龄正常值2s,并符合以下任意1项:①CD40L基因突变;②母系的表兄、舅舅或外甥确诊为XHIM。
3.2 可以诊断 男性,血清IgG水平≤相应年龄正常值2s,并符合以下所有条件:①T细胞数量正常,并对丝裂原有正常增殖反应;②B细胞数正常或增高,但缺乏抗原特异性IgG抗体;③以下1项或多项感染或合并症:生后5年内反复发生细菌感染,生后第1年发生卡氏肺囊虫感染,中性粒细胞减少症,隐孢子虫相关腹泻,硬化性胆管炎,微小病毒导致的再生障碍性贫血;④用可溶性CD40和抗CD40L单克隆抗体在活化的CD4+T细胞上不能标记出CD40L。
3.3 可能诊断 男性,血清IgG水平≤相应年龄正常值2s,T细胞和B细胞数量正常,并符合以下至少1项:①血清IgM水平≥相应年龄正常值2s;②生后第1年出现卡氏肺囊虫感染;③微小病毒导致的再生障碍性贫血;④隐孢子虫相关腹泻;⑤严重肝脏疾病(硬化性胆管炎)。
4 IgA缺乏症(IgAD)
4.1 明确诊断 >4岁患儿血清IgA水平<0.07 g·L-1,而血清IgG和IgM水平正常;排除其他导致低丙种球蛋白的病因;对疫苗有正常IgG抗体应答。
4.2 可以诊断 >4岁患儿血清IgA水平≤相应年龄正常IgA水平2s,而血清IgG、IgM水平正常;排除其他导致低丙种球蛋白的病因。对疫苗有正常IgG抗体应答。
5 选择性IgM缺乏症(SIgMD) 血清IgM<0.2 g·L-1,其他类别的免疫球蛋白水平正常。
6 重症联合免疫缺陷病(SCID)
6.1 明确诊断 <2岁患儿具有经胎盘传递而来的母体T细胞或CD3+T细胞<0.2,淋巴细胞绝对计数<3×109·L-1,并符合以下至少1项:①细胞因子共有的γ 链(γc)基因突变;②JAK3基因突变;③RAG1或RAG2基因突变;④IL-7Rα基因突变;⑤ADA活性低于对照的2%或其2个等位基因均突变。
6.2 可以诊断 <2岁患儿CD3+T细胞<0.2,淋巴细胞绝对计数<3×109·L-1,丝裂原增殖反应低于对照的10%或循环中出现母体淋巴细胞。本研究中无丝裂原增殖反应的记录,故<2岁患儿符合CD3+T细胞<0.2,绝对淋巴细胞计数<3×109·L-1,纳入可以诊断病例。
7 X连锁重症联合免疫缺陷病(XSCID)
7.1 明确诊断 男性,具有经胎盘传递而来的母体的T细胞或CD3+T细胞<0.1,CD16/56+NK细胞<0.02,CD19+B细胞>0.75,并符合以下任何1项:①细胞因子共同的γ链(γc)基因突变;②Northern blot检测淋巴细胞发现缺乏γc mRNA;③淋巴细胞或淋巴细胞系表面缺乏γc蛋白表达;④母系的表兄、舅舅或外甥患SCID。
7.2 可以诊断 男性,CD3+T细胞<0.1,CD16/56+NK细胞<0.02,CD19+B细胞>0.75,并符合以下所有表现:①生后第1年内生长发育停滞;②血清IgG、IgA水平≤相应年龄正常值2s;③持续或反复腹泻、上呼吸道感染或鹅口疮。
7.3 可能诊断 男性,外周血CD19+B细胞>0.4,并符合以下任何1项:①具有经胎盘传递而来的母体T细胞;②母系的表兄、舅舅或外甥有SCID病史。
8 WAS综合征
8.1 明确诊断 男性,患先天性血小板减少症(<70×109·L-1),血小板形态小,并且符合以下至少1项:①WASP基因突变;②Northern blot检测淋巴细胞发现缺乏WASP mRNA;③淋巴细胞缺乏WASP蛋白;④母系的表兄、舅舅或外甥血小板形态小,并患有血小板减少症。
8.2 可以诊断 男性,患先天性血小板减少症(<70×109·L-1),血小板形态小,并且符合以下至少1项:①湿疹;②对多糖抗原的抗体反应异常;③反复细菌或病毒感染;④自身免疫性疾病;⑤淋巴瘤、白血病或脑部肿瘤。
8.3 可能诊断 男性,患先天性血小板减少症(<70×109·L-1),血小板形态小,或男性患者因血小板减少症行脾切除术,且符合以下至少1项:①湿疹;②对多糖抗原抗体反应异常;③反复细菌或病毒感染;④自身免疫性疾病;⑤淋巴瘤、白血病或脑部肿瘤。
9 共济失调毛细血管扩张症(AT)
9.1 明确诊断ATM等位基因发生影响功能的突变,射线诱导后易发生染色体断裂,或患有进行性小脑共济失调。
9.2 可以诊断 发生进行性小脑共济失调,并符合以下3项:①眼部和面部毛细血管扩张;②血清IgA水平≤相应年龄正常值2s;③甲胎蛋白浓度≥相应年龄正常值2s;④射线诱导后易发生染色体断裂。
9.3 可能诊断 发生进行性小脑共济失调,并符合以下至少1项:①眼部和面部毛细血管扩张;②血清IgA浓度≤相应年龄正常值2s;③甲胎蛋白浓度≥相应年龄正常值2s;④射线诱导后易发生染色体断裂。
10 DiGeorge综合征
10.1 明确诊断 <3岁患儿CD3+T细胞降低(<0.5×109·L-1),并符合以下至少1项:①圆锥动脉干畸形合并实验室或临床诊断低钙血症;②圆锥动脉干畸形合并染色体22q11.2缺失;③实验室或临床诊断低钙血症合并染色体22q11.2缺失;④圆锥动脉干畸形, 实验室或临床诊断低钙血症,并有染色体22q11.2缺失。
10.2 可以诊断 <3岁患儿CD3+T细胞降低(<1.5×109·L-1),并有染色体22q11.2缺失。
10.3 可能诊断 <3岁患儿CD3+T细胞降低(<1.5×109·L-1),并符合以下至少1项:①心脏畸形;②实验室或临床诊断低钙血症;③面部畸形或上腭异常。
11 高IgE综合征(HIES) 依据美国国立卫生研究院(NIH)制定的HIES临床评分标准,总分>40分可临床诊断。
12 慢性肉芽肿病(CGD)
12.1 明确诊断 活化的中性粒细胞NBT或细胞呼吸爆发试验异常(低于对照的5%),并符合以下任意1项:①gp91、p22、p47或p67phox基因突变;②Northern blot检测缺乏上述任一基因的mRNA;③母系表兄、舅舅或外甥NBT或细胞呼吸爆发试验异常。
12.2 可以诊断 活化的中性粒细胞NBT或呼吸爆发试验异常(低于对照的5%),并符合以下任意1项:①葡萄球菌、黏质沙雷菌、念珠菌或曲霉菌所致深部位感染(肝脏、直肠周围或肺部脓肿、淋巴结炎、骨髓炎);②呼吸道、消化道和泌尿生殖道弥漫性肉芽肿;③生长发育停滞,肝、脾或淋巴结肿大。
13 X连锁淋巴组织增殖性疾病(XLP)
13.1 明确诊断 男性SH2D1A/SAP/DSHP基因突变,患有淋巴瘤/霍奇金病、致命EB病毒感染、免疫缺陷、再生障碍性贫血或淋巴细胞组织细胞疾病。
13.2 可以诊断 男性急性EB病毒感染危重症病例,患有淋巴瘤/霍奇金病、免疫缺陷、再生障碍性贫血或淋巴细胞组织细胞疾病,其母系的表兄、舅舅或外甥在急性EB病毒感染后也有过相似的诊断。
13.3 可能诊断 男性急性EB病毒感染危重症病例,患有淋巴瘤/霍奇金病、免疫缺陷、再生障碍性贫血或淋巴细胞组织细胞疾病。
14 Chediak-Higashi综合征(CHS)

15 家族性噬血细胞性淋巴组织细胞增生症(FLH)
出现持续发热,肝脾肿大,血细胞减少等临床表现,已知致病基因有PRF1、UNC13D、STX11和STXBP2等。
2013-08-17
2013-12-05)
张崇凡)
