枯否细胞在高脂诱导的肝脏胰岛素抵抗中的作用*
2013-12-03李树颖郭玉玺潘从清
李树颖 张 怡 郭玉玺 李 晶 潘从清
研究显示,胰岛素抵抗(insulin resistance,IR)是一种慢性炎症状态,巨噬细胞在肥胖脂肪组织的浸润、激活参与脂肪组织IR发生的观点已被接受[1],但枯否细胞(Kupffer cells,KCs)是否参与高脂诱导的肝脏IR还存在争议。本研究拟通过对不同阶段高脂喂养小鼠肝脏炎性因子表达的检测,观察其与肝脏IR发生的关系,探讨KCs在高脂诱导的肝脏IR中的作用。
1 材料与方法
1.1 实验动物 健康清洁级C57BL/6J小鼠84只,6周龄,体质量(15.62±3.14)g,雄性,购自中国医学科学院实验动物研究所。
1.2 方法
1.2.1 动物分组 按照随机数字表将小鼠随机分为普食组和高脂组,每组42只,每组内再随机分为7小组,每组6只。分别给予普食(脂肪占饲料热量组成的6.2%)和高脂(脂肪占饲料热量组成的59.3%)喂养。
1.2.2 血清胰岛素检测 6组普食和6组高脂小鼠分别在喂养第1、2、4、8、12和16周处死小鼠前空腹尾静脉取血,于4℃3 000 r/min离心10 min,取上清-80℃保存,按照小鼠胰岛素酶联免疫吸附(ELISA)试剂盒(shibayagi株式会社,日本)说明书操作检测小鼠血清胰岛素含量。
1.2.3 葡萄糖耐量试验 1组普食和1组高脂小鼠分别在普食和高脂喂养后1、2、4、8、12和16周行葡萄糖耐量检测,小鼠实验前一晚去除饲料,更换鼠笼、垫料,可自由饮用水,饥饿过夜16 h,次日晨轻剪鼠尾,血糖仪检测血糖后称质量,按1 g/kg腹腔注射葡萄糖溶液,分别在注射后15、30、60、120 min取血检测血糖。计算葡萄糖曲线下面积(AUG)=1/4×空腹值+1/2×15 min值+3/4×30 min值+60 min值+1/2×120 min值。
1.2.4 Western blot检测肝组织蛋白表达 分别在喂养第1、2、4、8、12和16周处死小鼠取肝脏,-80℃冻存。取肝组织0.1 g匀浆,RIPA细胞裂解液提取肝组织总蛋白,BSA法测定蛋白浓度。取60 μg蛋白样品经10%十二烷基磺酸钠-聚丙烯酰胺凝胶电泳(SDS-PAGE)分离后,转膜,封闭,免疫印迹法分别检测抗蛋白激酶B丝氨酸473(Akt-ser473)抗体、抗胰岛素受体底物1丝氨酸307(IRS1-ser307)抗体、抗核糖体蛋白S6激酶1苏氨酸389(S6K1-thr389)抗体、抗β肌动蛋白(βactin)抗体(均为1∶1 000稀释,Cell signaling公司,美国)或磷酸化c-Jun氨基末端激酶抗体(p-JNK,1∶1 000稀释,Santa公司,美国),4℃孵育过夜,羊抗兔IgG-HRP二抗(1∶5 000,Santa cruz公司,美国)室温孵育2 h,化学发光法显影,X线胶片压片曝光,用Image J软件处理,各蛋白的磷酸化相对表达量=磷酸化的蛋白水平灰度值/相对应β-actin灰度值。
1.2.5 单核细胞趋化蛋白(MCP)-1、肿瘤坏死因子(TNF)-α、白细胞介素(IL)-1β、IL-10 mRNA表达水平 Trizol提取肝组织总RNA。取2 μg总RNA,采用逆转录试剂盒(大连宝生物工程有限公司)合成cDNA,利用SYBR Green(罗氏公司,德国)在ABI7500实时定量-PCR仪进行扩增。引物序列β-actin 上游 5'-GGCTGTATTCCCCTCCATCG-3',下游 5'-CCAGTTGGTAACAATGCCATGT-3';MCP-1上游 5'-GCAGT⁃TAACGCCCCACTCA-3',下游 5'-CCCAGCCTACTCATT⁃GGGATCA-3';TNF-α上游5'-CGTCGTAGCAAACCACCAA-3',下游5'-GAGAACCTGGGAGTAGACAAGG-3';IL-1β上游 5'-ATCACTCATTGTGGCTGTGG-3',下游 5'-GTCGTTGCTTG⁃GTTCTCCT-3';IL-10 上游 5'-GCGTCGTGATTAGCGAT⁃GATG-3',下游 5'-CTCGAGCAAGTCTTTCAGTCC-3'。扩增反应采用25 μL体系:SYBR Green PCR混合物12.5 μL,上游引物 1 μL(10 μmol/L),下游引物1 μL(10 μmol/L),cDNA 5 μL,加双蒸水至25 μL。反应条件为:模板cDNA 94℃变性4 min后进行循环,变性94℃15 s,退火60℃10 s,延伸72℃10 s,共40个循环。记录目的基因Ct值和内参照基因Ct值,根据实时定量-PCR相对定量的2-△Ct法计算目的基因表达的相对值。
1.3 统计学方法 采用SPSS 12.0软件进行统计学分析,实验数据以x±s表示,两组比较采用t检验,Plt;0.05为差异有统计学意义。
2 结果
2.1 不同时间小鼠血清胰岛素水平比较 高脂喂养小鼠后1、2周,2组小鼠血清胰岛素差异无统计学意义,在第4、8、12和16周时,高脂组小鼠血清胰岛素水平均高于普食组(P<0.05或P<0.01),见表1。
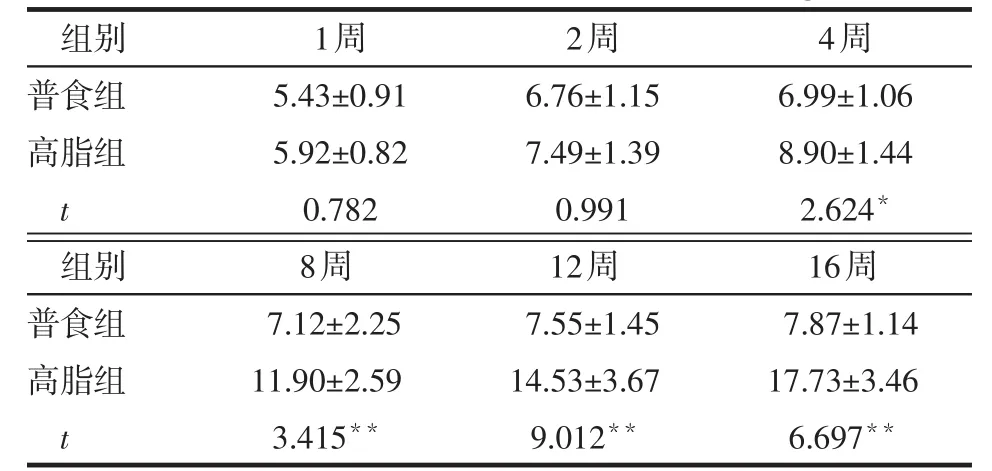
Table 1 Comparison of serum insulin levels in different time points between two groups表1 不同时间2组小鼠血清胰岛素水平比较(n=6,μg/L,x±s)
2.2 不同时间2组小鼠葡萄糖耐量曲线下面积比较 和普食组相比,高脂组1、2、4、8、12、16周的葡萄糖耐量曲线下面积均增大(P<0.05或P<0.01),见表2。
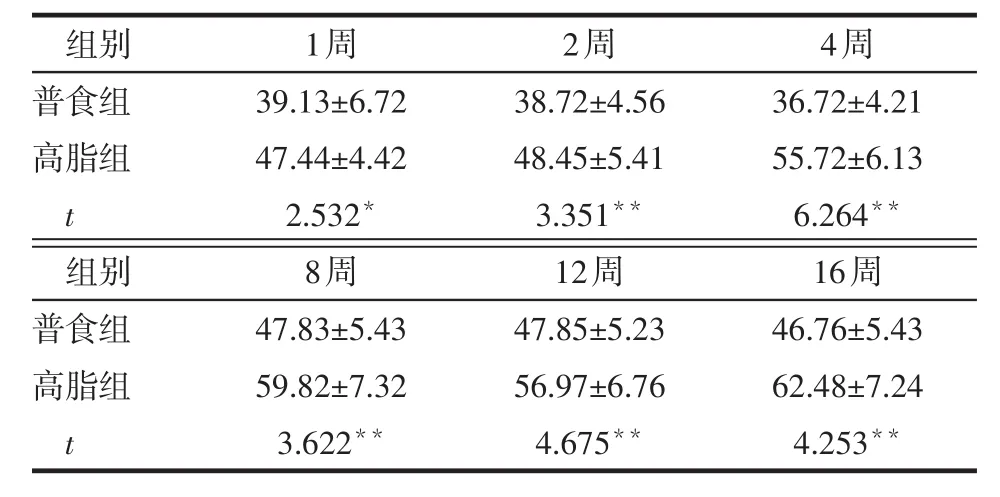
Table 2 Comparison of areas under the curve of glucose tolerance test between two groups表2 2组小鼠葡萄糖耐量试验曲线下面积比较(n=6,mmol·L-1·min-1,x±s)
2.3 不同时间2组小鼠肝脏IRS-1、S6K1、Akt、JNK磷酸化表达比较 与普食组相比,高脂组小鼠肝脏IRS1-ser307、S6K1-thr389(第4~16周)、p-JNK表达增强(2~16周),Akt-ser473(8~16周)表达减弱,见图1。1~2周时,2组小鼠IRS1-ser307、S6K1-thr389相对表达量差异无统计学意义(P>0.05),第4~16周高脂组高于普食组(均P<0.01),见表3、4。1~4周时,2组小鼠Akt-ser473相对表达量差异无统计学意义(均P>0.05),第8~16周时高脂组低于普食组(均P<0.01),见表5。第2~16周时,高脂组小鼠p-JNK相对表达量均高于普食组(均P<0.01),见表6。

Figure1 Comparison of phosphorylation state of hepatic IRS-1,S6K1,Akt and JNK expressions in different feeding time between two groups of mice图1 不同时间2组小鼠肝脏IRS-1、S6K1、Akt、JNK磷酸化表达比较
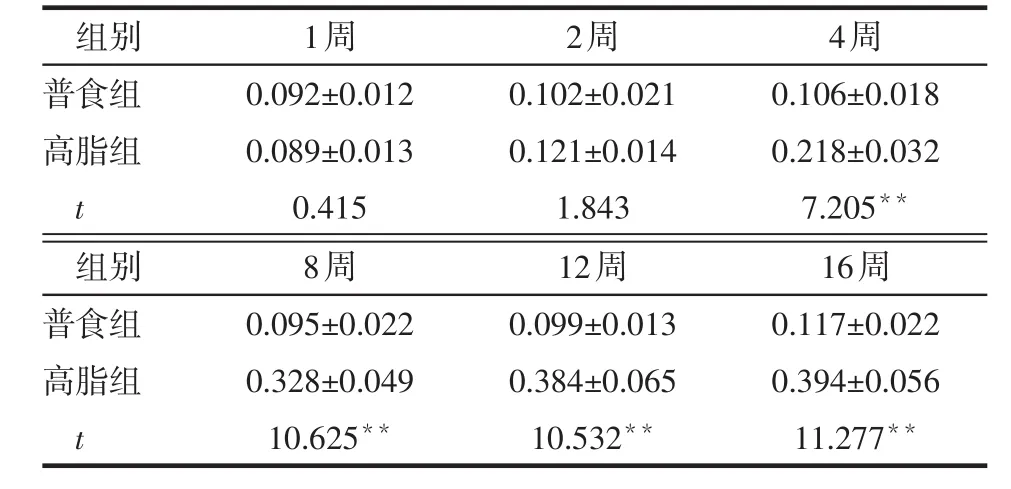
Table 3 Comparison of relative expression levels of hepatic IRS1-ser307 between two groups of mice表3 2组小鼠肝脏IRS1-ser307相对表达量比较(n=6,x±s)
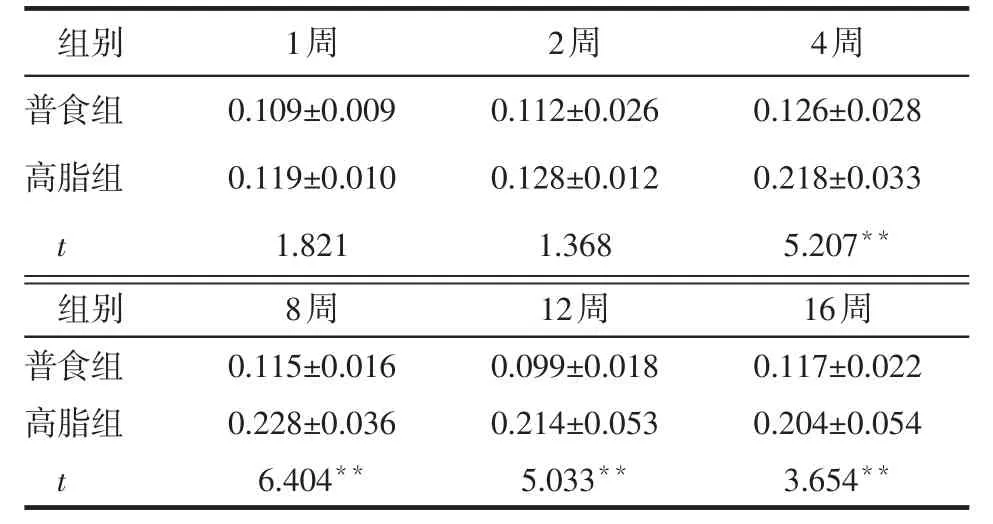
Table 4 Comparison of relative expression levels of hepatic S6K1-thr389 between two groups of mice表4 2组小鼠肝脏S6K1-thr389相对表达量比较(n=6,x±s)
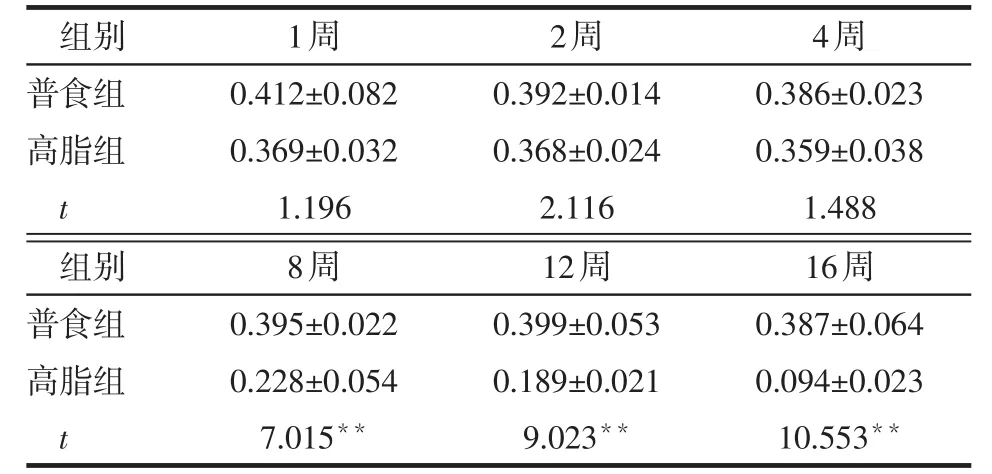
Table 5 Comparison of relative expression levels of hepatic Akt-ser473 between two groups of mice表5 2组小鼠肝脏Akt-ser473相对表达量比较(n=6,x±s)
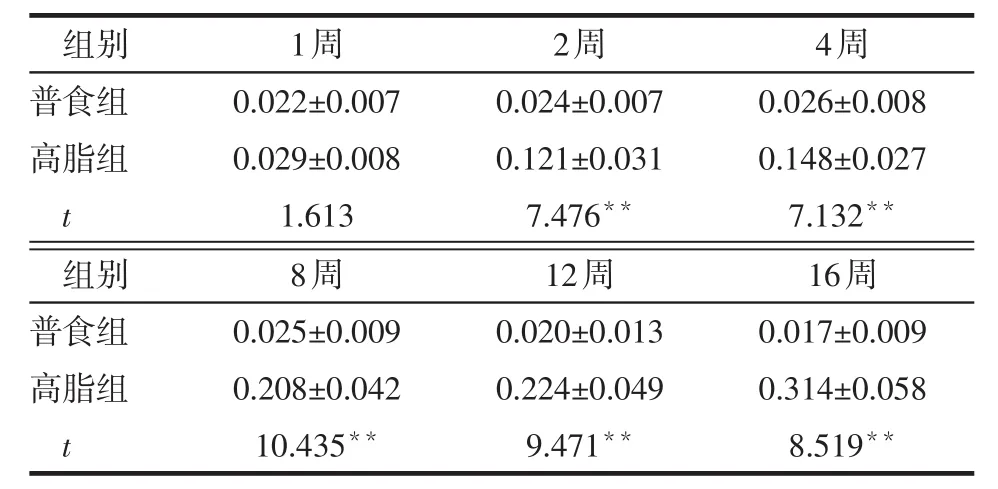
Table 6 Comparison of relative expression levels of hepatic p-JNK between two groups of mice表6 2组小鼠肝脏p-JNK相对表达量比较(n=6,x±s)
2.4 不同时间2组小鼠炎性因子表达变化 1周时,2组小鼠MCP-1、TNF-α、IL-1β mRNA表达比较差异无统计学意义(P>0.05),第2~16周时,高脂组上述指标表达量均高于普食组(均P<0.01),见表7~9。而第1~2周时2组抗炎因子IL-10 mRNA表达差异无统计学意义(P>0.05),第4~16周时,高脂组低于普食组(均P<0.01),见表10。
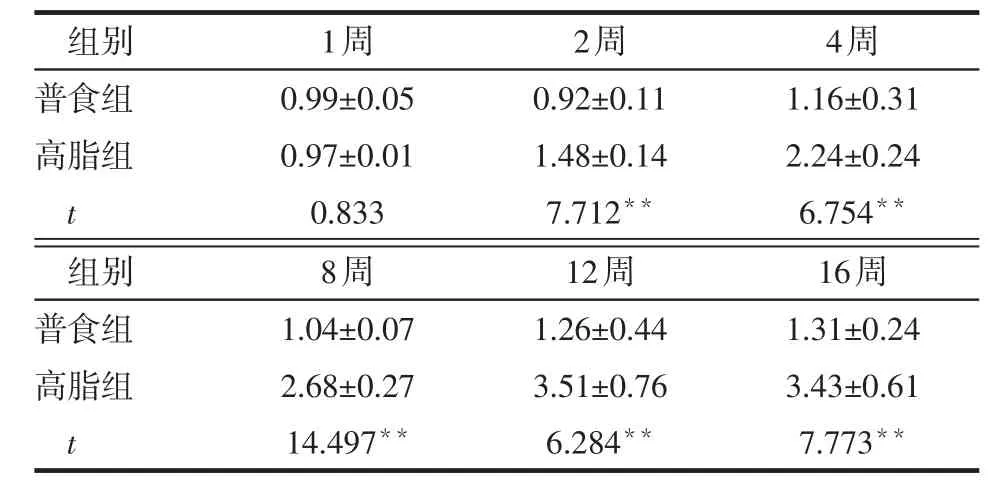
Table 7 Comparison of hepatic MCP-1 mRNA expression between two groups of mice表7 2组小鼠肝脏MCP-1 mRNA表达的比较(n=6,x±s)
3 讨论
3.1 巨噬细胞在IR中发挥作用 巨噬细胞可分为“传统”激活巨噬细胞(M1型)和“选择”激活巨噬细胞(M2型)2种,M1型主要分泌TNF-α、IL-1β等炎性因子;M2型主要分泌IL-10等抗炎因子,炎性因子可激活c-Jun氨基末端激酶(JNK)通路引起胰岛素受体底物(IRS)1丝氨酸磷酸化增加,导致胰岛素信号传导障碍[2];IL-10则通过抑制转录信号转导子与激活子3(STAT3)在胰岛素信号中发挥保护作用[3]。研究发现,高脂喂养小鼠1周,脂肪组织巨噬细胞(adipose tissue macrophage,ATM)即开始在脂肪组织浸润,且随喂养时间及肥胖增加而浸润加重[4],在此过程中ATM表型由抗炎的M2型向致炎的M1型转变,且与IR的程度一致[5]。KCs是肝脏巨噬细胞,其是否同ATM一样在肝脏IR中发挥作用目前还存在争议。
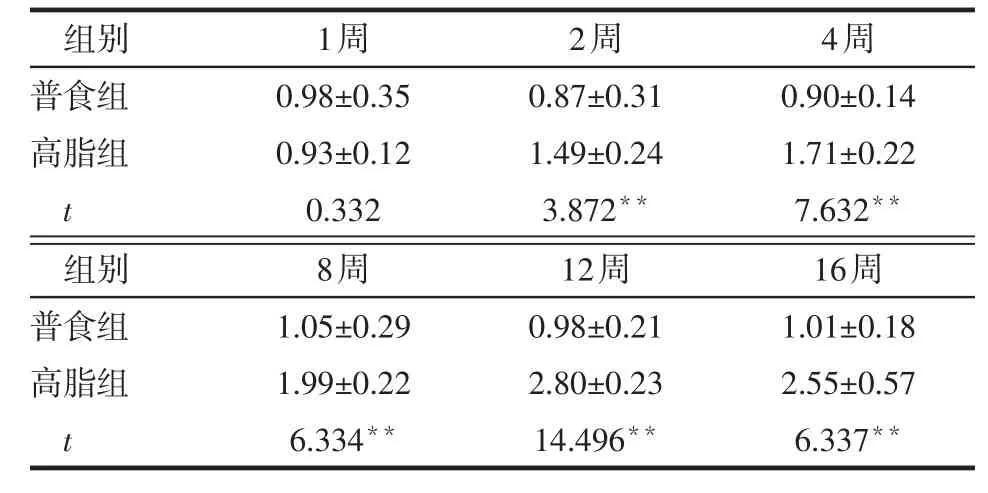
Table 8 Comparison of hepatic TNF-α mRNA expression between two groups of mice表8 2组小鼠肝脏TNF-α mRNA表达的比较(n=6,x±s)
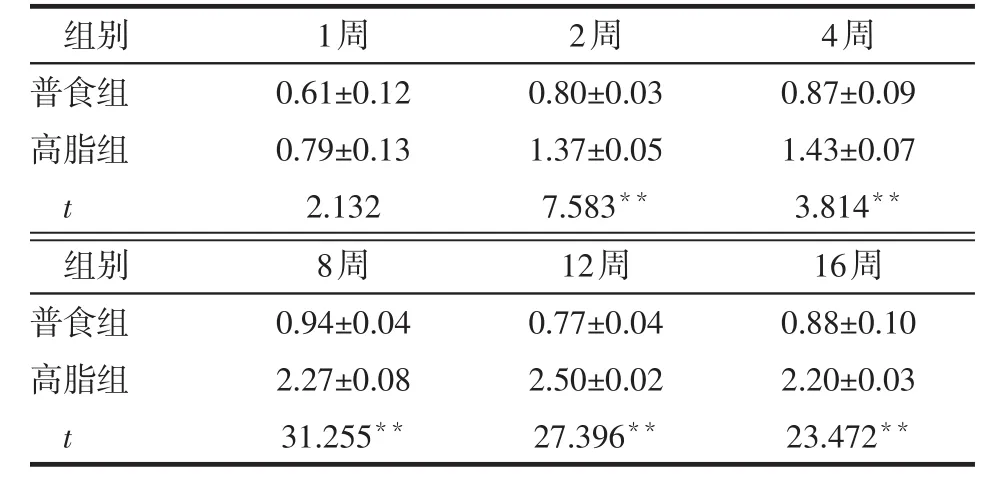
Table 9 Comparison of hepatic IL-1β mRNA expression between two groups of mice表9 2组小鼠肝脏IL-1β mRNA表达的比较(n=6,x±s)

Table 10 Comparison of hepatic IL-10 mRNA expression between two groups of mice表10 2组小鼠肝脏IL-10 mRNA表达的比较(n=6,x±s)
3.2 KCs参与肝脏IR的发生 本研究发现,高脂喂养小鼠第2~16周炎性因子TNF-α、IL-1β mRNA在肝脏表达高于普食组,抗炎因子IL-10 mRNA第4~16周表达低于普食组。Western blot结果也显示高脂饮食喂养第2周,高脂组小鼠肝脏炎性信号p-JNK相对表达量高于普食喂养小鼠,显示高脂饮食引起小鼠肝脏处于炎性状态。第4周开始,高脂组肝脏IRS1-307、S6K1-thr389相对表达量高于普食组;第8周高脂组Akt-ser473相对表达量开始低于普食组,提示出现肝脏IR。MCP-1可趋化循环中的单核细胞进入肝脏转变为巨噬细胞[6]。本研究发现在小鼠喂养第2周开始,高脂组MCP-1 mRNA表达开始高于普食组,推测高脂饮食可能通过征募单核细胞进入肝脏引起KCs在肝脏浸润增加,并促进肝脏M1型KCs释放炎性因子、抑制M2型KCs释放抗炎因子,通过影响肝脏胰岛素信号从而引起了肝脏IR的发生。另外,本研究发现高脂还可引起血清胰岛素水平增加,造成小鼠对葡萄糖代谢能力的下降,推测高脂造成的肝脏IR可引起肝糖原合成能力下降,从而在糖耐量异常中发挥一定作用。
3.3 饱和脂肪酸促进肝脏IR 近年来有研究认为KCs介导了高脂诱导的肝脏IR[7],但也有研究认为其保护肝脏免于高脂诱导的IR[8]。高脂饮食中的饱和脂肪酸成分可激活M1巨噬细胞介导IR[9],而单不饱和脂肪酸则激活M2巨噬细胞发挥保护作用[10],不同的研究结果可能与高脂中的饮食成分有关。本研究使用的高脂饮食以猪油为主,为饱和脂肪酸,故通过激活M1型KCs细胞、抑制M2型KCs介导肝脏IR的发生。
综上所述,在营养过剩条件下,高脂饮食可能通过调节M1型KCs分泌炎性因子TNF-α、IL-1β,抑制M2型KCs分泌抗炎因子IL-10在肝脏IR中发挥作用。但高脂是通过何种途径在肝脏诱导该改变的发生有待进一步研究。
[1]Lee J.Adipose tissue macrophages in the development of obesity-in⁃duced inflammation,insulin resistance and type 2 diabetes[J].Arch Pharm Res,2013,36(2):208-222.
[2]Copps KD,White MF.Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2[J].Diabetologia,2012,55(10):2565-2582.
[3]Gaba A,Grivennikov SI,Do MV,et al.Cutting edge:IL-10-mediat⁃ed tristetraprolin induction is part of a feedback loop that controls macrophage STAT3 activation and cytokine production[J].J Immu⁃nol,2012,189(5):2089-2093.
[4]Tacke F.Functional role of intrahepatic monocyte subsets for the pro⁃gression of liver inflammation and liver fibrosis in vivo[J].Fibrogen⁃esis Tissue Repair,2012,6(5):S27.
[5]Lynch L,Nowak M,Varghese B,et al.Adipose tissue invariant NKT cells protect against diet-induced obesity and metabolic disorder through regulatory cytokine production[J].Immunity,2012,37(3):574-587.
[6]Osborn O,Olefsky JM.The cellular and signaling networks linking the immune system and metabolism in disease[J].Nat med,2012,18(3):363-374.
[7]Clementi AH,Gaudy AM,van Rooijen N,et al.Loss of Kupffer cells in diet-induced obesity is associated with increased hepatic steatosis,STAT3 signaling,and further decreases in insulin signal⁃ing[J].Biochim Biophys Acta,2009,1792(11):1062-1072.
[8]Lanthier N,Molendi-Coste O,Horsmans Y,et al.Kupffer cell acti⁃vation is a causal factor for hepatic insulin resistance[J].Am J Physiol Gastrointest Liver Physiol,2010,298(1):107-116.
[9]Bhargava P,Lee CH.Role and function of macrophages in the meta⁃bolic syndrome[J].Biochem J,2012,442(2):253-262.
[10]Papackova Z,Palenickova E,Dankova H,et al.Kupffer cells amelio⁃rate hepatic insulin resistance induced by high-fat diet rich in monounsaturated fatty acids:the evidence for the involvement of al⁃ternatively activated macrophages[J].Nutr Metab(Lond),2012,22(3):9-22.
