炭气凝胶微球的物理活化与化学活化比较*
2013-11-24刘宁
刘 宁
(广东药学院医药化工学院,广东中山 528458)
炭材料由于具有成本低廉、性能稳定、原料来源广泛等优点,是目前最常用的超级电容器电极材料[1-9]。与传统炭材料相比,球形炭材料特殊几何形状使其具有独特的优势:球形结构完整,表面光滑,粒径均一,填充密度高,机械强度高等。炭气凝胶微球 (CA spheres)是近年来开发的新型球形炭材料[10-12],结合悬浮聚合与溶胶-凝胶法制备而成,其合成工艺成本低,安全系数高,制备与成型一次完成,更利于工业化应用。然而,在将炭气凝胶微球应用到超级电容器的电极材料的过程中,我们发现炭气凝胶微球的微观孔隙的结构不够理想,若应用于超级电容器其电化学性能仍有待提高。
为了丰富孔结构,提高电化学性能,研究者经常对材料进行活化和表面改性。材料的活化主要分为物理活化和化学活化。物理活化采用氧化性气体,如CO[13-15]2、水蒸气等[15-16],使石墨微晶中的炭部分气化,使炭材料内部形成新孔以及扩大原来的孔,从而形成发达的孔隙结构。化学活化是采用 KOH[17-19]、 ZnCl2[20-21]、 H3PO4等为活化剂[22-23],与炭质前驱体按一定比例混合,升温至600~1000℃,由于活化剂的脱水作用,原料中的氢和氧以水蒸气的形式释放,形成孔隙发达的材料。两种方法均能改善材料的孔隙结构,然而不同的方法对不同材料的孔结构和储电性能的影响存在差异,针对炭气凝胶微球的活化效果尚未有报道。
为提高炭气凝胶微球在电极材料方面的应用前景,改善炭气凝胶微球的微观孔结构,本文拟以炭气凝胶微球为原料,分别采用CO2和KOH作为活化剂,考察并对比两种活化方法对炭气凝胶微球孔结构和电化学性能的影响,探讨物理活化和化学活化对炭气凝胶微球结构性能的作用机制与差异。
1 实验
1.1 气凝胶微球的制备
一定配比的甲醛、间苯二酚、十六烷基三甲基溴化铵 (CTAB)和蒸馏水混合得到反应物溶液,将该溶液倾入装有花生油的三颈瓶中,在一定温度下搅拌进行悬浮聚合,液滴发生凝胶化反应就得到有机凝胶微球。将花生油过滤,所得微球转移到玻璃安瓶里,加少量蒸馏水后密封,在一定温度下老化5 d。老化后经洗涤的微球在室温下自然干燥1 d,然后在60℃左右的红外灯下烘1 d,最后放入100℃的烘箱中干燥3 h,得气凝胶微球。
1.2 气凝胶微球的活化
1.2.1 CO2活化 取一定量上述方法制得的气凝胶微球,在N2保护下,以5℃/min的速率从室温加热至活化温度 (活化温度分别为750℃和900℃),通入CO2气体,保持3 h。经自然冷却后得到CO2活化的炭气凝胶微球。
1.2.2 KOH活化 取一定量上述方法制得的气凝胶微球按50 mL/g的比例浸泡于浓度为φ=15%的KOH水溶液中,于30℃下振荡24 h。过滤,100℃下干燥12 h。干燥后的样品在N2保护下,以5℃/min的加热速率从室温加热到900℃,在此温度下分别保持1 h与3 h。经自然冷却后得到KOH活化的炭气凝胶微球。
1.3 活化后的炭气凝胶微球的结构表征
将活化前后的炭气凝胶微球贴于样品台上镀铂金,采用JSM-6330F型扫描电镜观察微球的微观孔结构。
采用VHX-1000C型超景深三维显微系统观察活化前后的炭气凝胶微球表面形貌。
采用ASAP2010吸附仪测定样品的比表面积并分析孔径结构。利用BET方法计算比表面积SBET,用t-plot法计算微孔表面积Smic和微孔体积Vmic,由BJH理论计算中孔的孔容VBJH和中孔表面积SBJH,按照DFT理论分析样品的孔径分布。
1.4 炭气凝胶微球电极的制备
将活化前后的炭气凝胶微球粉末与聚四氟乙烯按93∶7的质量比混合,加入少量电解液搅拌成膏状,均匀涂抹在泡沫镍上,制成约2 cm×1 cm的长方形电极,每片电极上的炭气凝胶的质量约为15 mg。电解液为φ=30%KOH溶液。
1.5 电化学测量与比电容的计算
循环伏安 (CV)、恒流充放与交流阻抗(EIS)均由IM6E型电化学工作站测定。循环伏安与交流阻抗采用三电极体系,以Ag/AgCl电极为参比电极、光滑的Pt片为辅助电极,φ=30%的KOH水溶液为电解液。CV法的扫描速度为50 mV/s,EIS法的测试频率范围为 10 kHz~100 mHz,电位控制±5 mV。
恒流充放法采用双电极体系制备模拟电容,电流控制为1 mA/cm2。
对电容器电容量的计算采用公式:
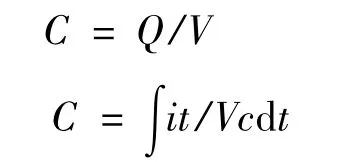
式中,i为充电电流;Vc为电容器两端的电压;t为电容器两端电压由0→Vc时所用的时间。以上数据的Vc均约为1 V。
式中,比电容是在2 mA的充电电流下测量并计算得到;m为炭气凝胶微球的质量。
2 结果与讨论
2.1 活化前后炭气凝胶微球的表观与微观形貌
为了表征炭气凝胶微球在活化前后的形貌变化,本文分别采用超景深三维显微镜与扫描电镜观察材料的外观形貌与微观结构。如图1的三维图片所示,经CO2或KOH活化后,炭气凝胶微球依然保持了圆球状外观。扫描电镜图 (如图2)也显示,经过活化后,微观结构基本与活化前一致,纳米粒子呈葡萄状堆叠,粒子与粒子之间包含大量孔隙。


2.2 活化对炭气凝胶微球孔结构的影响
为了观察不同活化方法对炭气凝胶微球孔结构的影响,经CO2和KOH在不同活化条件下得到的4种样品 (750℃下CO2活化的样品标记为CO2-750,900℃下CO2活化的样品为CO2-900;KOH活化1 h的样品为KOH-1 h,KOH活化3 h的样品为KOH-3 h)以及未被活化的原始样品 (标记为Original)通过N2吸附仪分析其内部微观孔结构,得到的N2吸附脱附等温线如图3所示。经CO2活化后,样品的吸附脱附曲线形状与未经活化的样品基本类似,滞后回环较为狭窄且在较高相对压力处出现,反映出典型的中孔材料特征;然而经KOH活化后,曲线形状明显改变,其滞留回环宽大许多,说明材料孔径分布较宽。同时还可以观察到,随着CO2活化温度的提高以及KOH活化时间的延长,不同样品的曲线在低相对压力下的部分有上升的趋势,说明随着活化程度的提高,活化赋予材料的微孔也在增加。
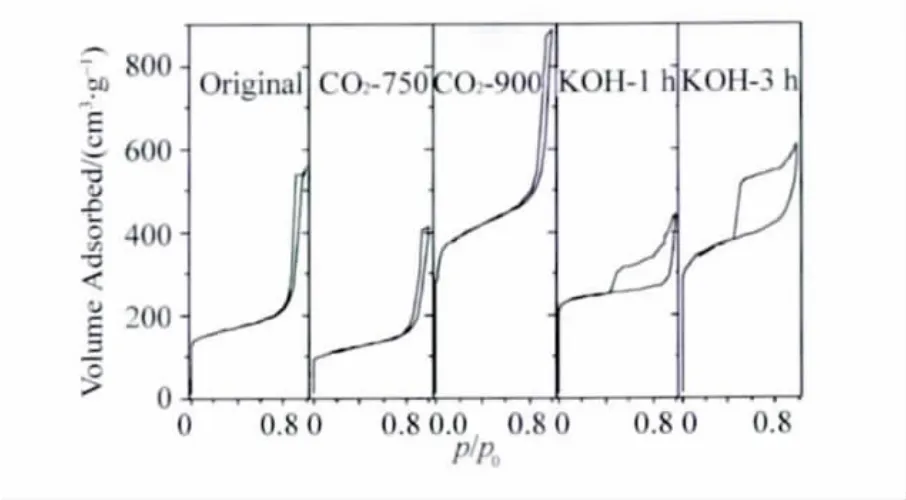
图3 活化前后炭气凝胶微球的氮吸附等温线Fig.3 N2adsorption-desorption isotherms of the CA spheres before and after activation
表1所列的详细孔结构参数进一步说明两种活化方法对炭气凝胶微球孔结构的改善的机理是不相同的。经过不同方法活化后,虽然材料的BET比表面积均有不同程度的增大趋势,然而这种趋势是来源于不同孔洞的贡献。从CO2活化的样品可以看出,活化后样品基本保持未活化前的中孔材料特征,中孔体积 (VBJH)较大,随着活化程度的提高,中孔体积还有增大的趋势。然而KOH活化的样品则不同,活化后样品的中孔体积减少,微孔体积 (Vmicro)显著增加,说明通过KOH活化,材料中部分中孔被破坏而坍塌,同时由于KOH良好的浸渍效果,能够较均匀地分散于微球中,从而提供更多的刻蚀点,更容易产生大量的微孔。对样品的孔径分布分析 (如图4所示)也证明了上述推断。经过活化,不论是CO2还是KOH,材料的孔径分布均变宽。然而不同的是,CO2活化后,中孔基本得到保持;KOH活化后,中孔迅速减少,微孔增多。
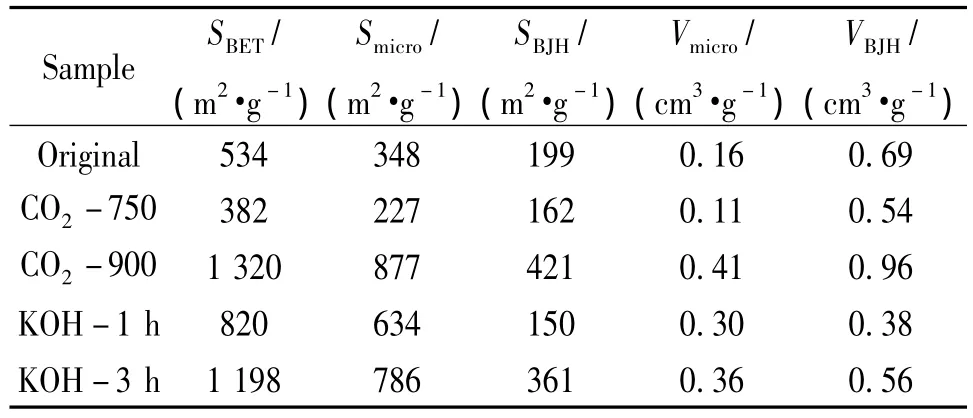
表1 活化前后炭气凝胶微球孔结构的变化Table 1 Pore structure changes of the CA spheres before and after activation

图4 活化前后炭气凝胶微球的孔径分布Fig.4 Pore size distributions of the CA spheres before and after activation
从表1还可以发现,通过CO2750℃以下活化时,材料中不论是中孔还是微孔,其面积、体积均有所下降,推测在此温度下,CO2与碳发生反应,破坏了部分的交联网络,导致材料内部不仅微孔还有部分的中孔都发生了塌陷。这样的活化对材料结构的改善是不利的。然而当活化温度达到900℃以上,材料的中孔、微孔的面积和体积均迅速增大。这说明,只有当活化程度足够充分时,CO2与碳的反应继续进行,使得碳不断被消耗,而形成新的微孔或者将原来的孔拓宽成为中孔。充分而深入的CO2活化更有利于炭气凝胶微球孔结构的改善。
2.3 活化对电化学性能的影响
孔结构分析表明,虽然CO2和KOH对炭气凝胶微球的活化机理是不一样的,但是均可以改善材料的孔结构,也必将对活化样品的电化学性能产生重要影响。活化前后的炭气凝胶微球样品被制作成电极,采用恒流充放、循环伏安与交流阻抗等方法测试了材料的电化学性能。
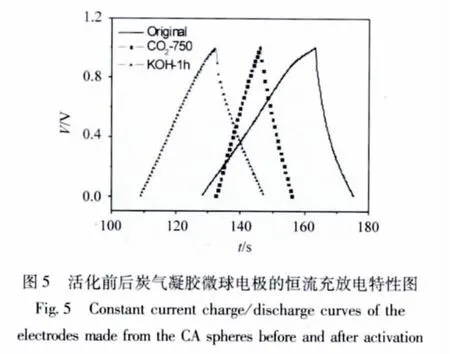
图5是活化前后炭气凝胶微球电极的恒流充放电曲线。由图可知,不论是CO2活化还是KOH活化,活化样品的充放电曲线均为近似直线,表明它们都具有较好的电容特性。通过恒流充放的结果计算,两种活化方法所制得的材料的比电容 (C)均得到不同程度的提高 (如图6所示),最高可为未活化样品的3倍,达247 F/g。由此可见,活化不仅可以改善炭气凝胶微球的孔结构,更有利于提高材料的电化学性能。
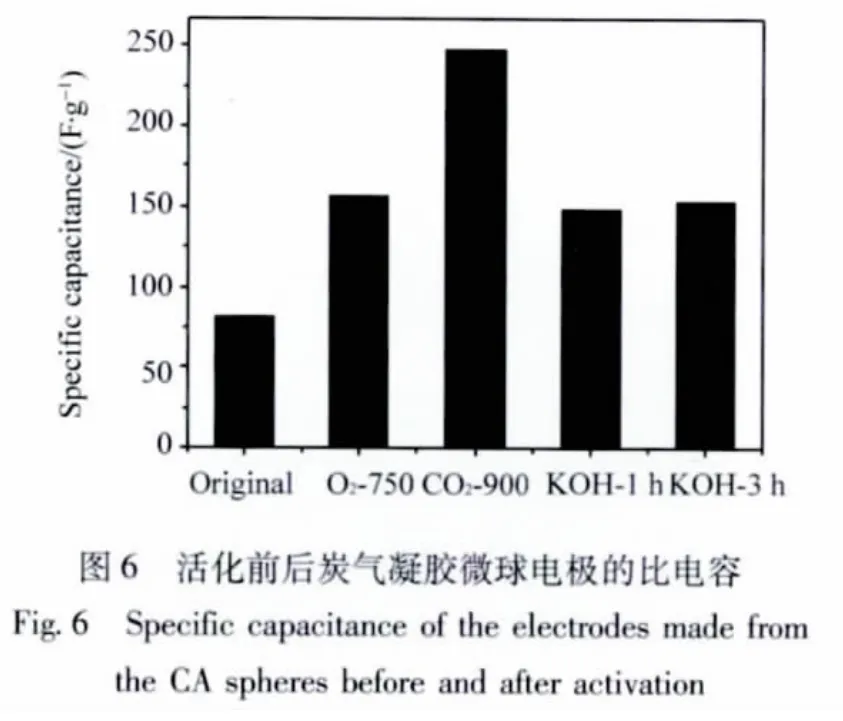
活化前后的炭气凝胶微球电极的循环伏安闭合曲线 (如图7所示)比较进一步说明了活化对电化学性能的改善作用。经过活化后,不论是CO2还是KOH,活化的循环曲线比未活化的曲线所包围的面积显著增大,证明了材料的比电容 (如图6数据)确实通过活化得到了提高。另外,活化后样品曲线在垂直方向的斜率明显增大,表明材料的电阻比未活化前减小。值得注意的是,KOH活化与CO2活化相比,则斜率相对较小一些,电阻应比其稍大一点。
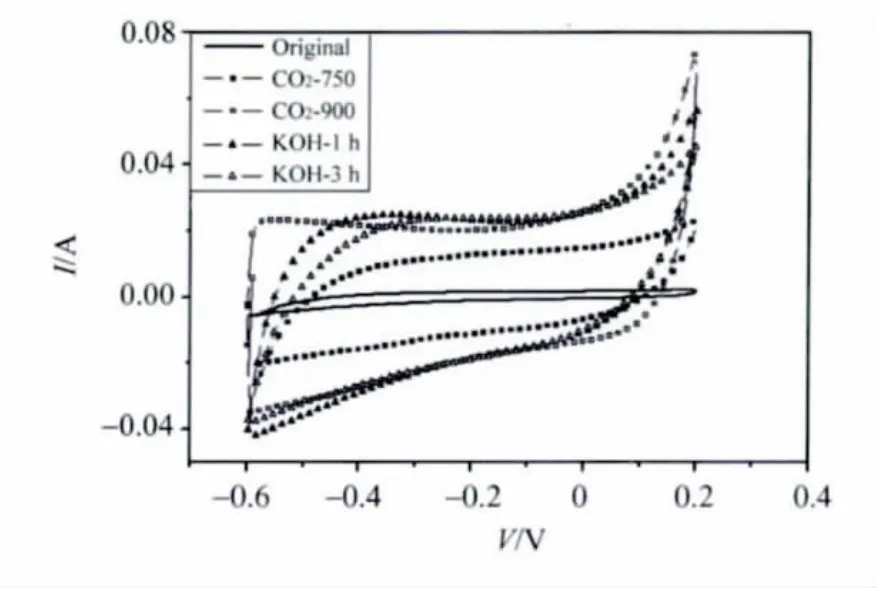
图7 活化前后炭气凝胶微球电极的循环伏安特性Fig.7 Voltammetry characteristics of the electrodes made from the CA spheres before and after activation
活化前后样品的交流阻抗曲线比较 (如图8所示)证明了这个分析。在低频率下,未活化炭气凝胶微球电极的阻抗曲线斜率较小,不符合理想电容的特征,反映此时电荷不是完全进入电极的孔,电解液对电极的浸润性差,很难进入材料中较小的孔内,表现为电极的内阻较大。活化后,材料阻抗曲线的低频部分斜率很大,近似竖直线,接近理想电容行为,说明材料经活化,电解液对电极浸润性好,反映为电阻较小。更值得注意的是,与CO2活化样品相比,KOH活化样品在高频区出现的圆弧明显较大,反映出材料的接触电阻也比较大,这与循环伏安曲线所反映的情况是一致的。结合两种活化对炭气凝胶微球孔结构与电化学性能的影响,可以推断,正是由于KOH活化后材料的部分中孔坍塌减少,使得电子进出的通道由原来的中孔变为孔径更小的微孔,进出变得困难,电子的运动速度降低,导致宏观表现为电阻增大。
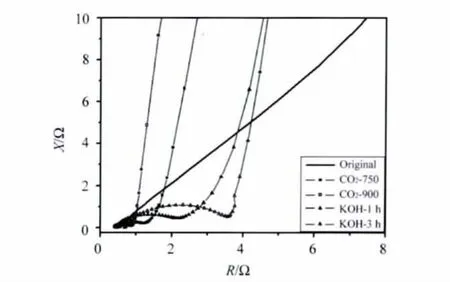
图8 活化前后炭气凝胶微球电极的阻抗曲线Fig.8 Nyquist plots of the electrodes made from the CA spheres before and after activation
3 结论
不论是以CO2为代表的物理活化还是以KOH为代表的化学活化,均能有效改善炭气凝胶微球的孔结构,活化后材料的比表面积最高可达1320 m2/g。然而,两种方法对炭气凝胶微球的活化机理有所不同,CO2活化保持了材料的中孔面积与体积,KOH活化则破坏了材料的中孔同时产生大量的微孔。
同时活化也对炭气凝胶微球的电化学性能产生重要影响。CO2和KOH活化均有效地提高了材料的电化学性能,活化后的比电容最高可为活化前的3倍,达247 F/g。虽然电容量均得到提高,但是CO2活化后的产品与KOH活化的产品相比,电阻更小一些。这反映出两种活化导致了不同的微观孔结构,CO2活化保持了大量的中孔,为电子的进出提供了快速通道,导致材料的电阻更小一些;而KOH活化虽然破坏了中孔,却同时产生了大量的微孔,增大了电化学活化表面,提高电化学性能。
通过活化,有利于炭气凝胶微球在超级电容器的电极材料方面的应用与推广。
[1]XIE K,QIN X T,WANG X Z,et al.Carbon nanocages as supercapacitor electrode materials[J].Adv Mater,2012,24(3):347-352.
[2]JHA N,RAMESH P,BEKYAROVA E,et al.High energy density supercapacitor based on a hybrid carbon nanotube reduced graphite oxide architecture[J].Adv Energy Mater,2012,2(4):438 -444.
[3]LIANG Y R,LI Z H,FU R W,et al.nanoporous carbons with a 3D nanonetwork-interconnected 2D ordered mesoporous structure for rapid mass transport[J].J Mater Chem:A,2013,1(11):3768-3773.
[4]LI S Y,WANG Y W,WU D C,et al.preparation and electrochemical properties of nitrogen enriched wormholelike nanoporous carbon[J].Adv Sci Let,2012,12(1):214-217.
[5]MCDONOUGH J K,FROLOV A I,PRESSER V,et al.Influence of the structure of carbon onions on their electrochemical performance in supercapacitor electrodes[J].Carbon,2012,50(9):3298-3309.
[6]BOUKHALFA S,EVANOFF K,YUSHIN G.Atomic layer deposition of vanadium oxide on carbon nanotubes for high-power supercapacitor electrodes[J].Energy Eviron Sci,2012,5(5):6872 -6879.
[7]FAN H S,WANG H,ZHAO N,et al.Hierarchical nanocomposite of polyaniline nanorods grown on the surface of carbon nanotubes for high-performance supercapacitor electrode[J].J Mater Chem,2012,22(6):2774-2780.
[8]TANG Z,TANG C H,GONG H.A high energy density asymmetric supercapacitor from nano-architectured Ni(OH)2/carbon nanotube electrodes[J].Adv Funct Mater,2012,22(6):1272 -1278.
[9]BOSE S,KUILA T,MISHRA A K,et al.Carbon-based nanostructured materials and their composites as supercapacitor electrodes[J].J Mater Chem,2012,22(3):767-784.
[10]LIU N,ZHANG S T,FU R W,et al.Carbon aerogel spheres prepared via alcohol supercritical drying[J].Carbon,2006,44(12):2430-2436.
[11]LIU N,ZHANG S T,FU R W,et al.The fabrication and structure of carbon aerogel spheres prepared by inverse suspension/emulsion polymerization and ambient pressure drying method[J].J Appl Polym Sci,2007,104(5):2849-2855.
[12]刘宁,符若文,王俊冰.炭气凝胶微球的储电性能研究[J].中山大学学报:自然科学版,2006,45(5):59-63.
[13]MACIA-AGULLO J A,MOORE B C,CZZORLA-AMOROS D,et al.Activation of coal tar pitch carbon fibres:Physical activation vs chemical activation[J].Carbon,2004,42(7):1367-1370.
[14]徐斌,吴峰,曹高萍,等.炭化温度对CO2活化PAN基活性炭纤维微结构的影响[J].新型炭材料,2006,21(1):14-18.
[15]RODRIGUEZ-REINOSO F,MOLINA-SABIO M,GONZALEZ M T.The use of steam and CO2as activating agents in the preparation of activated carbons[J].Carbon,1995,33(1):15-23.
[16]韩鹏献,王成扬,时志强,等.水蒸汽活化树脂炭用做双电层电容器电极材料[J].无机材料学报,2007,22(6):1046-1050.
[17]LU C L,XU S P,GAN Y X,et al.Effect of pre-carbonization of petroleum cokes on chemical activation process with KOH[J].Carbon,2005,43(11):2295-2301.
[18]付晓亭,贾凡,李文斌,等.马铃薯淀粉微孔炭微球的制备及其电化学性能[J].物理化学学报,2012,28(8):1906-1912.
[19]LI M,LI W,LIU S X.Hydrothermal synthesis,characterization,and KOHactivation of carbon spheres from glucose[J].Carbohydrate Research,2011,346(8):999-1004.
[20]ZHAI D Y,DU H D,LI B H,et al.Porous graphitic carbons prepared by combining chemical activation with catalytic graphitization[J].Carbon,2011,49(2):725-729.
[21]YUE Z R,CHRISTIAN L M,JAMES E.Preparation of fibrous porous materials by chemical activation:1.ZnCl2activation of polymer-coated fibers[J].Carbon,2002,40(8):1181-1191.
[22]LIOU T H.Development of mesoporous structure and high adsorption capacity of biomass-based activated carbon by phosphoric acid and zinc chloride activation[J].Chem Eng J,2010,158(2):129-142.
[23]JAGTOYEN M,DERBYSHIRE F.Activated carbons from yellow poplar and white oak by H3PO4activation[J].Carbon,1998,36(7/8):1085-1097.
