拉帕替尼中间体4-[3-氯-4-(3-氟苄氧基)苯氨基]-6-碘喹唑啉的合成新方法
2013-10-29贾玉才卢俊金郑彦军李宝林
贾玉才,卢俊金,郑彦军,李宝林
(教育部药用资源及天然药物化学重点实验室,陕西师范大学 化学化工学院,陕西 西安710062)
拉帕替尼(lapatinib)是英国葛兰素史克生产的一种口服小分子酪氨酸激酶抑制剂,2007年在美国上市,用于肿瘤的临床治疗,为4-苯氨基喹唑啉类衍生物.它是靶向作用于表皮生长因子受体HER1和HER2的双重酪氨酸激酶抑制剂[1],可逆性的作用于酪氨酸激酶ATP结合位点,通过抑制酪氨酸激酶的磷酸化而抑制肿瘤细胞的生长与繁殖[2].
Petrov等[3]报道的拉帕替尼及其衍生物的合成中以4-氯-6-碘喹唑啉为原料经与3-氯-4-(3-氟苄氧基)苯胺间的取代反应得中间体1.在这个过程中使用的4-氯-6-碘喹唑啉需用高毒性、高污染的氯化试剂氯化亚砜或三氯氧磷来制备,后处理困难,对环境污染较大.季兴等[4]以2-氨基-5-碘苯甲腈(5)在N,N-二甲基甲酰胺二甲缩醛(DMF-DMA)中回流缩合成N′-(2-氰基-4-碘苯基)-N,N-二甲基甲脒(6),而后与3-氯-4-(3-氟苄氧基)苯胺一起在乙酸中回流,经Dimroth重排得到中间体1,虽然该方法革除了氯代试剂的使用,但邻氨基苯甲腈(4)碘代反应仍用到高毒、腐蚀性大,而且在光照和空气中不稳定的氯化碘,产率也不高.本文报道拉帕替尼合成中所需重要中间体4-[3-氯-4-(3-氟苄氧基)苯氨基]-6-碘喹唑啉(1)的一种合成新方法.该方法在总结该中间体以往合成方法的基础上,对其不足之处进行了改进,通过氰基化[5]、还原、碘代[6]、甲脒生成和Dimroth重排[7]五步反应得到拉帕替尼中间体1,总收率52.2%.
1 实验
1.1 主要仪器与试剂
GCMS-QP2010E(岛津国际贸易有限公司)[GCMS条件:色谱柱Rxi-5ms柱(30m×0.25 mm×0.25μm),柱温310℃,载气He,氦气流量1.04mL/min;GC条件:色谱柱RxiR-1ms柱(30m×0.25mm×0.25μm),柱温280℃,载气He,氦气流量1.00mL/min];超导傅立叶数字化核磁共振谱仪(Bruker Avance,300MHz,400MHz);XT—5A显微熔点测定仪(北京市科仪电光仪器厂).
邻硝基苯甲醛,安庆长江医药化工有限公司;2-氯-4-硝基苯酚,南京康满林生物医药科技有限公司;间氟苄基氯,北京市大田丰拓化学技术有限公司;盐酸羟胺,天津市恒兴化学试剂制造有限公司.3-氯-4-(3-氟苄氧基)苯胺按文献[8-9]方法自制.
1.2 合成
1.2.1 化合物3的合成 取化合物2(45.34g,0.30mol)和甲酸钠(40.80g,0.60mol)溶于甲酸(120mL)中,在100~105℃将盐酸羟胺(25.72g,0.37mol)分6次于1h内加入,用TLC跟踪(展开剂:乙酸乙酯-石油醚(1∶2))反应至完毕,约需反应时间5.5~6h.将反应混合物冷至室温,抽滤.滤液旋蒸回收甲酸,所得残留物与滤饼混合,悬浮于200 mL饱和食盐水中搅拌30min.抽滤,滤饼水洗、干燥得黄色化合物3(26.90g,90.8%),m.p.111~113℃ (文 献 [10]:收 率90%),1H NMR(300 MHz,CDCl3)δ:7.84(m,2H),7.94(t,1H),8.36(t,1H).GC-MS(m/z):148[M]+.
1.2.2 化合物4的合成 取化合物3(4.44g,0.03mol)、保险粉(16.70g,0.10mol)、CH2Cl2(45 mL)置于250mL三颈瓶中,在40℃下滴加水(90 mL),反应30~40min后TLC跟踪[展开剂:乙酸乙酯-石油醚(1∶2)]发现原料已消耗完,冷却(有不溶物滤去),分液,CH2Cl2相干燥、蒸除溶剂.水相在40℃下滴加浓盐酸(18mL),滴加完毕搅拌1h.冷却至室温,用20%NaOH水溶液调pH值至9—11,用乙酸乙酯萃取.萃取液经干燥、蒸除溶剂.两次旋干后的产物合并可得到黄绿色固体化合物4(2.48g,70%),m.p.49~51℃(文献[11]:m.p.48~50℃),1H NMR(300MHz,CDCl3)δ:4.39(s,2H),7.40(t,2H),7.30-7.40(m,2H).GCMS(m/z):118[M]+.
1.2.3 化合物5的合成 取化合物4(0.59g,5 mmol)、NH4I(0.78g,5.4mmol)、HOAc(10mL)置于50mL三颈瓶中,在20℃下搅拌30min后,滴加30%H2O2(3.74g,33mmol),滴完后继续在20℃下搅拌,96h后反应完毕.向体系中加入亚硫酸钠(1.24g)的水溶液(10mL),再在搅拌下滴加20%NaOH水溶液使反应混合物至中性.搅拌30 min后抽滤,水洗、干燥,得棕黄色固体产物化合物5(1.13g,92.6%),m.p.88~90℃(文献[4]:收率84.5%),1H NMR(300MHz,CDCl3)δ:4.46(s,2H),6.53(d,J=8.1Hz,1H),7.56(d,J=8.1Hz,1H),7.65(s,1H).GC-MS(m/z):244[M]+.
1.2.4 化合物6的合成 取硫酸二甲酯(0.38g,3mmol)和N,N-二甲基甲酰胺(0.22g,3mmol)置于25mL的圆底烧瓶中,70℃下搅拌3h,即生成亚胺盐.
取化合物5(0.12g,0.50mmol)溶于甲苯(2 mL)中,室温下将其滴入上述亚胺盐中,搅拌10 min后加入甲醇钠(0.09g,1.74mmol),室温下继续搅拌9h,减压蒸出甲苯,向残留物中加入水(10 mL),用2mol/L硫酸水溶液调pH至3,搅拌30 min后用二氯甲烷(10mL)萃取.水相用20%NaOH水溶液调节pH至10,用CH2Cl2萃取(2×10mL),合并萃取液,无水硫酸镁干燥,浓缩得黄色固体化合物6(0.13g,89.3%),m.p.54~55℃,1H NMR(300MHz,CDCl3)δ:7.77(s,1H),7.65(d,J=8.5Hz,1H),7.59(s,1H),6.70(d,J=8.5Hz,1H),3.08(s,6H).GC-MS(m/z):299[M]+.
1.2.5 化合物1的合成 取化合物6(1.46g,5 mmol)、3-氯-4-(3-氟苄氧基)苯胺 (1.33g,5.30 mmol)和乙酸(3mL)置于50mL三颈瓶中.在130℃下搅拌10min体系出现黄色固体,冷却至室温,减压抽滤.滤饼用水悬浮,氨水调节pH至10,搅拌1h.抽滤,滤饼用水洗至中性,干燥,得灰白色固体化合物1(2.51g,99.3%),m.p.228~229℃(文献[4]:收率82.4%),1H NMR(400MHz,DMSO-d6)δ:5.26(s,2H),7.15-7.22(m,1H),7.29-7.35(m,3H),7.44-7.51(m,1H),7.56(d,J=11.6Hz,1H),7.75(dd,J=12.0,3.4Hz,1H),8.03(d,J=3.4Hz,1H),8.11(dd,J=11.6,2.2Hz,1H),8.61(s,1H),8.95(d,J=2.2Hz,1H),9.85(s,1H,NH).
2 结果与讨论
现有拉帕替尼制备中所需中间体化合物1的合成方法存在一些缺点,我们对其不足进行了改进,实验所用的合成路线如图1所示.
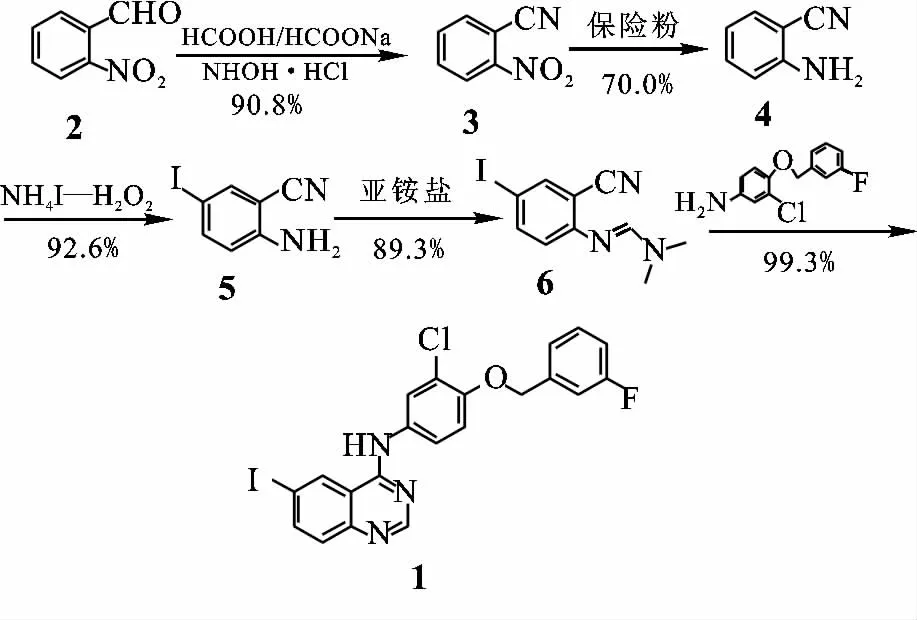
图1 化合物1的合成路线Fig.1 Synthetic route of compound 1
在按照该路线进行化合物1合成的过程中取得了两方面的改进,一是在化合物5的制备中采用NH4I-H2O2体系进行碘代;二是利用硫酸二甲酯与N,N-二甲基甲酰胺生成的亚胺盐直接制备化合物6.
2.1 2-氨基-5-碘苯甲腈(5)的制备
在已报道的路线中化合物4的碘代多用氯化碘作为碘代剂,它的缺点是高毒、腐蚀性大,而且在光照和空气中不易保存.为避免这种不足,实验改用绿色环保的NH4I-H2O2体系进行碘代.在双氧水的作用下,碘化铵中的碘负离子被氧化后与乙酸作用生成CH3COO-I+,生成物与化合物4通过芳环上的亲电取代反应得到化合物5,过程如图2所示.
起初以TLC对反应跟踪的结果表明,补加碘化铵和改变温度对反应均没有改善,而增加双氧水的量以及延长反应时间对反应影响较大.因此,我们借助气相色谱仪对双氧水用量及反应时间的影响进行跟踪及反应条件的优化,结果如表1所示.当双氧水物质的量6倍于化合物4,反应96h时为最佳的反应条件,此时由GC分析计算的收率达100%.由于双氧水的存在会使部分的碘离子被氧化为碘单质,以黑色晶体的状态吸附在瓶壁上,用1mol/L的亚硫酸钠溶液处理,3mol/L NaOH溶液调pH至中性后,再经抽滤、水洗、干燥就可以得到产物化合物5,分离收率为92.6%.
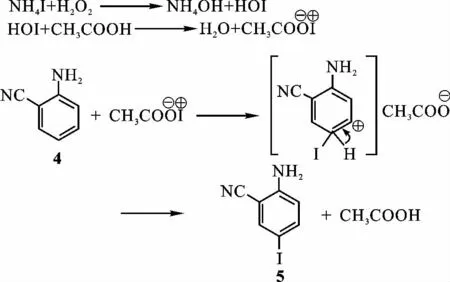
图2 NH4I-H2O2体系的一种可能的碘代反应机理Fig.2 A possible mechanism for the iodination of NH4I-H2O2system

表1 双氧水与2-氨基苯甲腈摩尔比和反应时间对反应收率的影响*Tab.1 Effect of the mole ratio of H2O2to 2-anthranilonitrile and reaction time on the reaction yield
2.2 N′-(2-氰基-4-碘苯基)-N,N-二甲基甲脒(6)的制备
文献[4]报道化合物6的制备方法是用DMFDMA与化合物5反应得到,实际上用到的商品DMF-DMA仍是由硫酸二甲酯和DMF在甲醇钠的存在下制备得到,且收率不高[12].本实验由硫酸二甲酯和DMF反应生成的亚胺盐在甲醇钠的存在下直接与化合物5作用得到化合物6(反应如图3),省去了DMF-DMA制备、分离过程.然后,化合物6再与3-氯-4-(3-氟苄氧基)苯胺经Dimroth重排反应得到所需中间体化合物1.

图3 化合物6的合成Fig.3 Synthetic route of compound 6
在制备化合物6的实验条件优化中,发现反应溶剂以甲苯最佳,当换甲醇为溶剂时,反应几乎不进行.亚胺盐与化合物5的投料配比对反应结果有较大的影响,具体如表2所示.当亚胺盐与化合物5的摩尔比由4∶1提高到6∶1时,化合物6收率呈增长的趋势;摩尔比由6∶1增大到12∶1的过程中,收率呈下降趋势.当二者的摩尔比为6∶1时,收率达到最高为89.3%.后处理中,减压蒸出甲苯后,加水,再用2mol/L硫酸水溶液调pH至3.此时化合物6会以盐的形式溶在水溶液中,以二氯甲烷萃取除掉不溶性有机物后,再将得到的水相用20%NaOH水溶液调节pH至10,产物便游离出来.再用CH2Cl2萃取、干燥,蒸除溶剂可得到纯的化合物6.
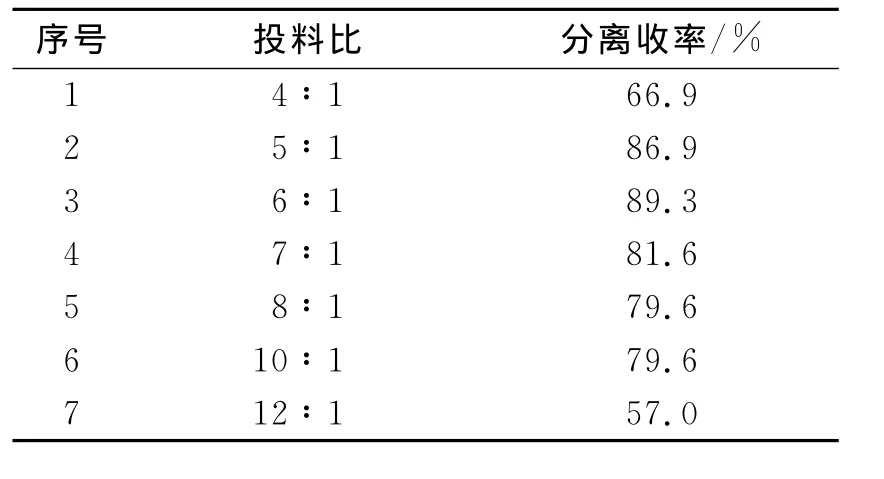
表2 投料比对反应收率的影响Tab.2 Effect of feed ratio on the reaction yield
3 结论
本文通过对文献报道的化合物1合成路线的改进,得到较为环保的该化合物合成新方法.在化合物4的碘代反应中,采用NH4I-H2O2体系的绿色碘代方法,代替了环境不友好的氯化碘的使用,使所得方法具有操作简单,后处理方便的优点.在合成化合物6一步反应中,由硫酸二甲酯和DMF生成的亚胺盐直接与芳胺成脒,省去了DMF-DMA的制备过程,降低了生产成本,同时也提高了产率.本文报道的拉帕替尼中间体化合物1合成路线较以往的方法更为绿色环保,操作更简单,每步的收率均较高,五步反应总收率为52.2%.
[1]Morphy R.Selectively nonselective kinase inhibition:striking the right balance[J].Journal of Medicinal Chemistry,2010,53(4):1413-1437.
[2]Rana P,Sridhar S S.Efficacy and tolerability of lapatinib in the management of breast cancer[J].Breast Cancer:Basic and Clinical Research,2012,6:67-77.
[3]Petrov K G,Zhang Yuemei,Carter M,et al.Optimization and SAR for dual ErbB-1/ErbB-2tyrosine kinase inhibition in the 6-furanylquinazoline series[J].Bioorganic &Medicinal Chemistry Letters,2006,16(17):4686-4691.
[4]季兴,王武伟,许贯虹,等.拉帕替尼的合成[J].中国医药工业杂志,2009,40(11):801-804.
[5]Xiong Xiaodong,Chen Wenxue,Kuang Yunyan,et al.A novel and practical synthesis of 2-Amino-5-hydroxypropiophenone[J].Organic Preparations and Procedures International:The New Journal for Organic Synthesis,2009,41(5):423-427.
[6]Narender N,Reddy K S K,Krishna Mohan K V V,et al.Eco-friendly oxyiodination of aromatic compounds using ammonium iodide and hydrogen peroxide[J].Tetrahedron Letters,2007,48(35):6124-6128.
[7]Jyothi P,Adibhatla K S,Venkaiah C.A new process for the preparation of lapatinib and its pharmaceutically acceptable salts:WO,2011039759[P].2011-04-07.
[8]Tang Pengcho,Feng Jun,Feng Feng,et al.Preparation methods of quinazoline derivatives and their pharmaceutical uses:WO,2009012647[P].2009-01-29.
[9]邹永,张学景,林慧贞.紫檀芪的合成方法:中国,1269782C[P].2006-08-16.
[10]Madhusudana Reddy M B,Pasha M A.Environment friendly protocol for the synthesis of nitriles from aldehydes[J].Chinese Chemical Letters,2010,21(9):1025-1028.
[11]Musso H,Schroder H.Mechanism of the oxygen transfer in the hydrogenation of 2-nitrobenzonitrile to 2-aminobenzamide[J].Chemische Berichte,1965,98(5):562-76.
[12]张海峰,李媛,王昭煜.N,N-二甲基甲酰胺二甲基缩醛的制备[J].化学试剂,1990,12(2):124-125.
