用于燃料电池Co@Pt/C 核壳结构催化剂的制备及表征
2013-09-21曹春晖赵天天马建新
曹春晖 林 瑞 赵天天 黄 真 马建新
(同济大学新能源汽车工程中心,上海201804;2同济大学汽车学院,上海201804)
1 引言
质子交换膜燃料电池(PEMFC)因具备能量转换效率高、可靠性高、无污染、组装和维修方便等优点而成为交通工具的理想替代动力系统之一.1但制约燃料电池大规模应用的两大障碍,即成本和耐久性问题尚未完全解决.质子交换膜燃料电池常用的催化剂是负载于炭黑的铂(Pt),它具有活性高、化学稳定性好的特点.2而Pt的储量却极为有限,稀缺性决定了其成本会随着燃料电池汽车的推广而急剧上升.因此,提高铂的利用率、降低铂的载量显得尤为重要.
在纳米尺度上对金属催化剂颗粒的结构进行理性设计和化学裁剪可显著改变金属催化剂的物理化学性质,获得性能更好的催化剂.其中核壳结构(记为“核@壳”)纳米金属颗粒具有特殊的电子结构及表面性质,因而在催化等领域的应用日益受到重视.3研究证明,以Pt为壳层的M@Pt(M为Co,Ni,Cu等过渡金属)核壳型催化剂对氧还原具有更高的催化活性.一方面,Pt壳层可防止过渡金属的溶解,防止电催化活性的衰减,增加Pt的比表面积,提高Pt的利用率;另一方面,核壳催化剂的核/壳之间可产生相互作用,添加过渡金属引起Pt氧还原反应(ORR)催化性能提高的机理主要是:减小了Pt-Pt之间的距离(几何效应),形成了具有更多5d轨道空位的电子结构,导致氧的π电子向Pt表面转移的增加(电子效应).4核壳结构燃料电池催化剂的研究和应用已有大量的报道,5-14其中Co@Pt催化剂表现出较高ORR活性.
有报道9通过置换和沉积方法成功地制备出负载碳的Pt-Co核壳结构催化剂,发现该催化剂的ORR活性是Pt/C催化剂的2-4倍.Wu等13用还原法制备了Co@Pt催化剂,通过X射线衍射(XRD)、透射电子显微镜(TEM)和电化学测试技术证明了核壳结构的形成.在电化学测试中该催化剂表现出优于Pt/C催化剂的ORR活性.Lee等14用热分解和后续还原方法制备了负载碳的Co@Pt催化剂,并考察了酸洗对催化剂性能的影响,发现Co@Pt具有良好的ORR催化剂性能,且经过适当酸处理的Co@Pt催化剂表现出更好的ORR活性.但目前对Co@Pt催化剂应用于燃料电池并且其耐久性的考察较少,因此深入地研究其结构、性能及耐久性显得很有必要.
本研究采用两步化学还原法制备了系列Co@Pt/C催化剂,用透射电镜(TEM)、X射线光电子能谱(XPS)分析以及电化学技术对其进行形貌和电化学性能、ORR动力学特性进行表征.考察了催化剂在电解质溶液中的稳定性及耐久性.
2 实验部分
2.1 Co@Pt/C催化剂的制备
实验所用的碳载体为XC-72碳黑(美国Cabot公司,比表面积为235 m2·g-1).氯化钴(CoCl2)、氯铂酸(H2PtCl6)、氢氧化钠(NaOH)、乙醇(C2H5OH)、乙二醇(EG)、聚乙烯吡咯烷酮 (PVP)、盐酸(HCl)、硝酸(HNO3)和异丙醇(C3H8O)均为分析纯,国药集团化学试剂有限公司;质量分数为5%的Nafion溶液购自美国Dupont公司.
碳黑的预处理:XC-72碳黑用2.0 mol·L-1盐酸在120°C回流处理4 h,以去除硫等无机杂质;用5.0 mol·L-1硝酸在120°C回流进行表面氧化处理4 h,以增加表面的官能团;过滤,用去离子水洗涤,100°C真空干燥12 h.
本研究采用两步还原法制备Co@Pt/C催化剂.第一步,通过还原CoCl2·6H2O制备Co粒子.15在0.1 mol·L-1CoCl2乙醇溶液中加入10 mL PVP乙醇溶液作为稳定剂,搅拌条件下缓慢加入新制备的0.05 mol·L-1NaBH4(97%)乙醇溶液到上述混合溶液中.在制备过程中通入氮气,以防止还原出的Co纳米粒子被氧化.加入NaBH4乙醇溶液反应30 min后,对产物用去离子水离心水洗4-5次,得到Co纳米粒子备用.
第二步,采用乙二醇还原H2PtCl6和Co的混合悬浮液制备Co@Pt/C催化剂.将一定量的XC-72碳黑载体分散在30 mL乙二醇中,超声振荡2 h备用.上一步制备的Co粒子分散在20 mL乙二醇中,溶液移入三颈烧瓶中,搅拌条件下加入一定量PVP作为稳定剂,搅拌30 min后逐滴加入0.03862 mol·L-1H2PtCl6乙二醇溶液.混合均匀后,用 2 mol·L-1NaOH乙二醇溶液将混合液pH值调为12.升温到120°C,回流搅拌反应2 h后加入制备好的碳浆,再于120°C下搅拌1h后,让其自然冷却.上述过程中均以氮气为保护气.待悬浮液温度降至室温后,加入5 mol·L-1的盐酸调节pH值到3.过滤所得产物,用大量去离子水清洗,于80°C真空干燥8 h,即制得Co@Pt/C催化剂.以同样的方法制备了Co/C粒子作为对比.为进一步还原Pt和Co及去除残留的稳定剂,对制得的催化剂在氢气气氛(5%H2+95%N2,40 mL·s-1)、300 °C条件下进行2 h还原处理.文中凡经过还原处理的催化剂标记为Co@Pt/C(reduced),未经还原处理的催化剂标记为Co@Pt/C.在制备的催化剂中,金属(Pt+Co)质量占催化剂总质量的40%.Pt和Co的原子比分别为3:1和1:1.
2.2 催化剂结构表征
采用JEM-2010透射电子显微镜(日本电子光学公司)对催化剂的金属粒子晶体大小、颗粒度和颗粒分布进行高分辨透射电子显微镜观察.加速电压为200 kV,采用铜网微栅样品架,样品在无水乙醇中超声分散,然后负载到样品架上制成电镜样品.
催化剂表面元素通过PHI Model 5700型X光电子能谱仪(美国PHI公司)进行分析.辐射源为Mg Kα(hν=1253.6 eV),电子结合能用碳(C 1s=284.6 eV)校正.
2.3 催化剂电化学表征
催化剂的电催化性能评价在上海辰华CHI104P电化学工作站上进行,采用美国PINE公司的AFMS-LXF型旋转圆盘电极(RDE)及装置.测试条件为0.1 mol·L-1HClO4的电解质,温度25 °C.参比电极为可逆氢电极(RHE),采用铂丝作为对电极,涂有催化剂层的玻碳电极为工作电极.电极催化层的制备过程如下:将2.0 mg催化剂加入5 mL称量瓶中,然后加入1 mL质量比为1:30的5%Nafion溶液和甲醇溶液,混合后超声30 min.分散均匀后用微量进样器抽取混合液10 μL,滴加到玻碳电极表面,干燥后进行测试.在电化学测试中,涂覆在电极表面上的催化剂质量相等.
循环伏安(CV)测试的扫描范围为0.05-1.15 V(vs RHE,下同),扫描速率为50 mV·s-1.在进行CV测试之前,HClO4溶液中先通N2半小时,以驱赶出溶液中的O2.计算CV曲线中氢吸脱附峰的面积,原子量级平滑的Pt催化剂表面的吸收电荷为210 μC·cm-2,据此可以推算出被测催化剂中Pt的电化学活性面积(ECSA).16为测试催化剂的耐久性,对催化剂进行500次CV循环扫描,实验条件同上.
线性伏安测试(LSV)的扫描范围为0.05-1.2 V(vs RHE,下同),扫描速率为5 mV·s-1,转速为1600 r·min-1.LSV扫描之前,HClO4溶液中通O2半小时,以使溶液中达到O2饱和.为计算ORR的动力学参数,分别在 300、600、800、1200、1600、2000和2400 r·min-1下进行LSV扫描,扫描范围为0.05-1.2 V,扫描速率为5 mV·s-1.
3 结果与讨论
3.1HR-TEM测试
图1所示是未经热还原和经过热还原的Co@Pt(1:3)/C催化剂的高分辨TEM(HR-TEM)照片.可以看出,催化剂颗粒在碳载体上分布均匀,这说明该制备方法可以得到分散均匀、粒径较小的催化剂颗粒.图中显示明显的亮度对比,表明在碳载体上附有不同的元素.可在图片中辨别出Co@Pt(1:3)/C催化剂中的Pt和Co.此外,从图可以清楚地看到,经过热还原处理后催化剂的粒径有所增大,晶格更加明显.未经热处理的催化剂粒径在2.5 nm左右,而经过300°C还原热处理后的催化剂粒径有所增大,至3.0 nm左右.
3.2 XPS测试
为了分析制备样品中元素价态分布,对制备的样品做了XPS测试,结果如图2所示.从图中可以清晰地观察到分别对应于Co 2p,Pt 4f,C 1s和O 1s的四个光电子峰.
图 2(a)是 Co@Pt(1:1)/C(reduced)和 Co@Pt(1:3)/C(reduced)催化剂的XPS全谱,图2(b)、图2(c)、图2(d)和图2(e)分别是催化剂的Pt(4f)和Co(2p)的XPS谱图.4f亚层电子自旋轨道耦合引起能级分裂而产生两个不同的峰,分别反映出4f5/2和4f7/2的简并度,峰的相对强度比为0.75.17通过XPS图谱,计算Co@Pt(1:1)/C(reduced)和 Co@Pt(3:1)/C(reduced)催化剂的4f5/2和4f7/2峰相对强度比分别为0.84和0.92.4f5/2和4f7/2峰的相对强度比偏离标准值的原因可能是由于有其他物质的存在,如添加的Co元素、载体碳,使其自旋耦合效应受到影响.

图1 Co@Pt(1:3)/C(reduced)和Co@Pt(1:3)/C催化剂的HR-TEM图Fig.1 HR-TEM images of Co@Pt(1:3)/C(reduced)and Co@Pt(1:3)/C electrocatalyst

图2 Co@Pt(1:1)/C(reduced)和Co@Pt(1:3)/C(reduced)系列催化剂的XPS图谱Fig.2 XPS spectra for as-prepared Co@Pt(1:1)/C(reduced)and Co@Pt(1:3)/C(reduced)catalysts
根据标准XPS谱,17Pt(0)和Co(0)的结合能分别是71.2、74.5 eV和778.3、793.7 eV.实验所制备的催化剂样品中Pt(0)所对应的结合能的值有所增大,表明其中部分Pt以Pt氧化物的形态存在.可能是空气中的氧气对Pt纳米粒子氧化所致.对XPS结果的Pt 4f峰进行拟合,其中的最强峰可归属于零价Pt,第二强峰可以归属为2价Pt的存在(Pt(II),PtO和Pt(OH)2).可以看出Pt 4f7/2和Pt 4f5/2的峰最为明显,表明在这两种催化剂上的Pt主要以还原态形式存在.
在XPS全谱图中也发现了对应于Co(0)2p的较弱的峰.通过XPS数据,可以计算得到Co@Pt(1:1)/C(reduced)催化剂中Pt和Co的含量分别为1.63%和0.24%,而在Co@Pt(1:3)/C(reduced)催化剂中该比例分别为1.75%和0.19%.同时也发现Co(0)2p峰变弱的同时,Pt(0)4f的峰增强.这些结果表明,催化剂表面的Co大部分被制备过程中沉积的Pt所包覆.
3.3 电化学性能测试
Co@Pt/C和商业Pt/C催化剂的电化学活性通过CV和LSV技术表征.催化剂的CV测试在氮气气氛、0.1 mol·L-1HClO4中进行,同时也进行了Co/C粒子的CV测试.测试结果如图3所示.氢吸脱峰出现在0-0.3 V,氧的还原氧化峰位于0.7-0.8 V之间,氢吸附峰的形状与商业Pt/C催化剂类似.而对于Co/C粒子,基本没有氢吸脱附峰出现.通过氢吸脱附峰,可以计算得到Pt/C、Co@Pt(1:3)/C(reduced),Co@Pt(1:1)/C(reduced),Co@Pt(1:3)/C和Co@Pt(1:1)/C催化剂的ECSA分别是52.2、53.1、50.4、45.2和29.6 m2·g-1.其中 Co@Pt(1:3)/C(reduced)催化剂的ECSA稍高于商用催化剂Pt/C(JM),可见Co@Pt核壳结构的形成有助于Pt利用率的提高.
从计算结果中也可以看出,催化剂中Co和Pt的比例对其性能有很大影响,通过调节该比例能使性能得到明显的提高.Co:Pt为1:1时,热还原前后其ECSA分别为29.6和45.2 m2·g-1,当该比例变为1:3时,热还原前后催化剂ECSA分别提高至为50.4和53.1 m2·g-1.
为研究催化剂的ORR性能,对其进行LSV测试,结果如图4所示.Co@Pt/C催化剂的ORR性能很大程度上受Co和Pt比例的影响,Co@Pt(1:3)/C(reduced)催化剂表现出最好的ORR性能,甚至要优于Pt/C(JM)催化剂.相比于Pt/C催化剂,Co@Pt(1:3)/C(reduced)催化剂LSV曲线半波电位有8 mV的正移.

图3 商用Pt/C(JM)和Co@Pt/C催化剂的CV曲线Fig.3 Cyclic voltammogram(CV)curves of as-prepared Co@Pt/C and commercial Pt/C(JM)catalysts
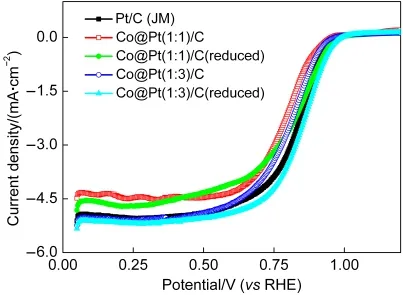
图4 制备的Co@Pt/C和Pt/C(JM)催化剂的线性伏安扫描(LSV)曲线Fig.4 LSV curves of O2reduction on Co@Pt/C and commercial Pt/C(JM)catalysts
Co@Pt(1:3)/C(reduced)催化剂ORR性能的提升可能是因为形成的核壳结构诱导产生了几何效应,这使得在颗粒表面出现更多的Pt催化活性位.此外,催化剂Pt-Pt原子间距也可能有所变化,这也导致其吸附和还原氧的能力.18有文献通过XRD数据计算得到的Pt-Co催化剂晶格常数要小于纯Pt的,Pt-Pt相邻原子之间的距离的减小被认为有助于提升催化剂的ORR性能.19,20Co@Pt(1:3)/C催化剂LSV曲线的半波电位为0.803 V,经过热还原处理后,Co@Pt(1:3)/C(reduced)催化剂的半波电位有所提升,达到0.842 V.通过LSV曲线可以发现,热处理之后的催化剂具有更好的ORR性能.制备的系列催化剂的ORR性能的排序为Co@Pt(1:3)/C(reduced)>Pt/C(JM)>Co@Pt(1:3)/C.
Co@Pt/C催化剂的ORR动力学性质的测试结果如图5所示.将Co@Pt(1:3)/C(reduced)涂覆在RDE上,在O2饱和的0.1 mol·L-1HClO4溶液中,进行LSV测试.氧还原的动力学性质用Koutecký-Levich关系式分析,分别以图5中0.45、0.60和0.75-1V下得到的电流倒数和转速平方根的倒数为对应关系作图,结果如图6所示,得到一组斜率相近的平行直线.
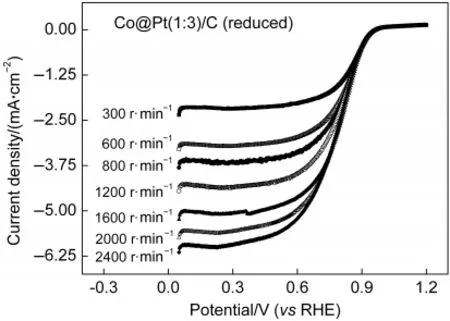
图5 不同转速下Co@Pt(1:3)/C(reduced)在0.1 mol·L-1 HClO4溶液中氧化还原性质的RDE测试结果Fig.5 Rotation disk electrade(RDE)measurements for ORR on Co@Pt(1:3)/C(reduced)in 0.1 mol·L-1HClO4 with different speeds
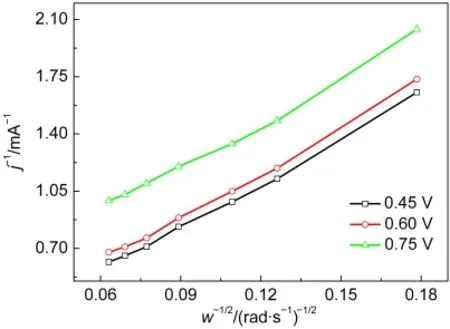
图6 Co@Pt(1:3)/C(reduced)催化剂氧还原反应的Koutecký-Levich图线Fig.6 Koutecký-Levich plots for ORR on Co@Pt(1:3)/C(reduced)catalyst
以下方程中,21斜率(1/B)直接与氧还原反应的总电子数相关联:
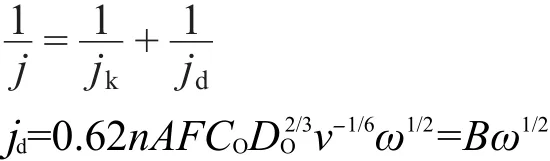
式中,j是实验观察到的电流,jk是动力学电流,n是每摩尔O2被还原时所转移的电子数目,F是法拉第常数,A是电极面积,DO(1.97×10-5cm2·s-1)是氧气的扩散系数,CO(1.2×10-6mol·cm-3)是氧气的溶解度,v(1.0×10-2cm2·s-1)是溶液的动力学粘度.在电压0.45-0.75 V之间,计算得到n值为4.09-4.19(表1),表明Co@Pt/C催化剂表面发生的ORR反应是以四电子路线进行的.
催化剂的稳定性与耐久性也是本实验研究的一个重要方面.将Pt/C(JM)催化剂和Co@Pt(1:3)/C(reduced)催化剂按实验部分描述的方法涂覆于工作电极,在0.1 mol·L-1HClO4溶液中进行500个循环的CV测试以考察其耐久性.实验结果如图7所示.在开始的100个CV循环后,催化剂性能几乎没有下降;但此后催化剂的性能有明显的下降.经过500个循环后,Co@Pt(1:3)/C(reduced)催化剂的ECSA下降了30.8%.而对于Pt/C催化剂,在开始的100个循环结束后,催化剂也基本保持了较高的性能,完成300个循环之后出现了一定的衰减,但与自制催化剂相比其衰减速率较小.500个循环之后,Pt/C催化剂的ECSA的衰减率为15.5%.可见自制催化剂的稳定性不如Pt/C(JM)催化剂,但是与文献22中报道的Pt-Co合金催化剂相比,稳定性有很大的提高.这可能是由于Pt包覆在过渡金属Co的外层,有效地阻止了Co的溶解和流失,从而提高了催化剂的稳定性和耐久性.可见,核壳结构是提高催化剂稳定性和耐久性的有效途径之一.
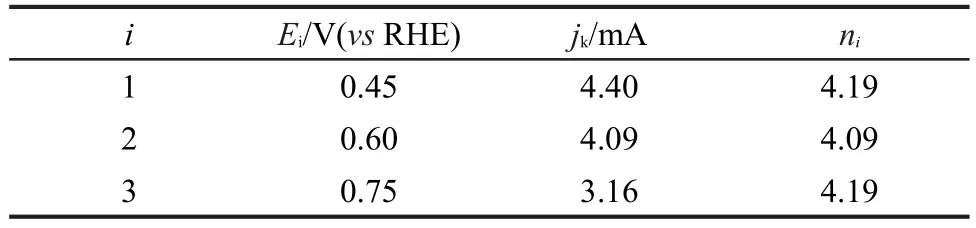
表1 在0.1 mol·L-1HClO4中不同电位下Co@Pt(1:3)/C(reduced)催化剂ORR动力学电流(jk)和电子转移数(ni)Table 1 Kinetic current(jk)and average number of(ni)electrons transferred for O2reduction reaction(ORR)on Co@Pt(1:3)/C(reduced)at different potentials in 0.1 mol·L-1HClO4
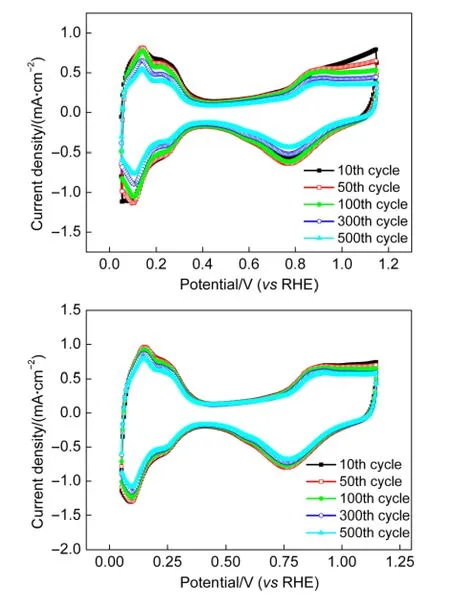
图7 室温氮饱和气氛下Co@Pt(1:3)/C(reduced)催化剂在0.1 mol·L-1HClO4电解质溶液中的耐久性Fig.7 Durability of as-prepared Co@Pt/C in N2-saturated 0.1 mol·L-1HClO4at room temperature
4 结论
通过HR-TEM表征结果可以看出本研究制备的Co@Pt/C催化剂形成了核壳结构,催化剂的粒径主要分布在3-5 nm范围内,热处理对催化剂的形貌也有较大的影响.通过XPS分析发现,纳米颗粒表现出Co 2p、Pt 4f、C 1s和O 1s四个明显的光电子峰,Pt主要是以金属态存在.催化剂电化学性能测试结果表明所形成的核壳结构在降低Pt用量的同时也提高了Pt的利用率.核壳结构的形成同时使得催化剂的稳定性和耐久性得到提升,制备得到的Co@Pt/C催化剂经溶液中500个循环的CV测试之后性能虽有所下降,但衰减程度远小于Pt-Co合金催化剂.此外,本研究进行了催化剂的动力学性质研究,发现催化剂ORR遵循四电子路线进行.
(1)Wang,Y.;Chen,K.S.;Mishler,J.;Cho,S.C.;Adroher,X.C.Appl.Energy 2011,88,981.doi:10.1016/j.apenergy.2010.09.030
(2)Wee,J.H.;Lee,K.Y.;Kim,S.H.J.Power Sources 2007,165,667.doi:10.1016/j.jpowsour.2006.12.051
(3) Zhang,H.Y.;Cao,C.H.;Zhao,J.;Lin,R.;Ma,J.X.Chin.J.Catal.2012,33,229.[张海艳,曹春晖,赵 健,林 瑞,马建新.催化学报,2012,33,229.]
(4) Carrette,L.;Friedrich,K.A.;Stimming,U.Fuel Cells 2001,1,15.
(5) Zhang,Z.L.;Yuan,J.N.;Sun,Y.P.;Liu,S.B.;Duan,D.H.;Hao,X.G.Chin.J.Inorg.Chem.2011,27,2413.[张忠林,员娟宁,孙彦平,刘世斌,段东红,郝晓刚.无机化学学报,2011,27,2413.]
(6) Gao,H.L.;Liao,S.J.;Zeng,J.H.;Liang,Z.X.;Xie,Y.C.Acta Phys.-Chim.Sin.2010,26,3193.[高海丽,廖世军,曾建皇,梁振兴,谢义淳.物理化学学报,2010,26,3193.]doi:10.3866/PKU.WHXB20101214
(7)Wang,G.X.;Wu,H.M.;Wexler,D.;Liu,H.K.;Savadogo,O.J.Alloy.Compd.2010,503,L1.
(8) Zhu,H.;Li,X.W.;Wang,F.H.Int.J.Hydrog.Energy 2011,36,9151.doi:10.1016/j.ijhydene.2011.04.224
(9)Kristian,N.;Yu,Y.L.;Lee,J.M.;Liu,X.W.;Wang,X.Electrochim.Acta 2010,56,1000.doi:10.1016/j.electacta.2010.09.073
(10) Choi,I.;Ahn,S.H.;Kim,J.J.;Kwon,O.J.Appl.Catal.B:Environ.2011,102,608.doi:10.1016/j.apcatb.2010.12.047
(11) Dang,D.;Gao,H.L.;Peng,L.J.;Su,Y.L.;Liao,S.J.;Wang,Y.Acta Phys.-Chim.Sin.2011,27,2379.[党 岱,高海丽,彭良进,苏允兰,廖世军,王 晔.物理化学学报,2011,27,2379.]doi:10.3866/PKU.WHXB20110922
(12) Sun,D.;He,J.P.;Zhou,J.H.;Wang,T.;Di,Z.Y.;Ding,X.C.Acta Phys.-Chim.Sin.2010,26,1219.[孙 盾,何建平,周建华,王 涛,狄志勇,丁晓春.物理化学学报,2010,26,1219.]doi:10.3866/PKU.WHXB20100507
(13)Wu,H.M.;Wexler,D.;Wang,G.X.;Liu,H.K.J.Solid State Electrochem.2012,16,1105.doi:10.1007/s10008-011-1486-5
(14) Lee,M.H.;Wang,P.S.;Do,J.S.J.Solid State Electrochem.2008,12,879.doi:10.1007/s10008-007-0477-z
(15)Liu,S.B.;Yuan,J.N.;Zhang,Z.L.;Duan,D.H.;Li,Y.B.;Hao,X.G.Chin.J.Inorg.Chem.2010,26,1171.[刘世斌,员娟宁,张忠林,段东红,李一兵,郝晓刚.无机化学学报,2010,26,1171.]
(16) Ryan,O'H.;Che,S.Y.;Whitney,C.;Fritz,B.P.Fuel Cell Fundamentals;Publishing House of Electronics Industry:Beijing,2007;p 196;translated by Wang,X.H.,Huang,H.[Ryan,O'H.,车硕源,Whitney,C.,Fritz,B.P.燃料电池基础.王晓红,黄 宏,译.北京:电子工业出版社,2007:196.]
(17) Moulder,J.F.;Stickle,W.F.;Sobol,P.E.;Bomben,K.D.Handbook of X-ray Photo-electron Spectroscopy;Perkin-Elmer Corporation:Minnesota,1992;pp 186-187.
(18) Santiago,E.I.;Varanda,L.C.;Villullas,M.J.Phys.Chem.C 2007,111,3146.doi:10.1021/jp0670081
(19) Kiros,Y.J.Electrochem.Soc.1996,143,2152.doi:10.1149/1.1836974
(20) Xiong,L.;Manthiram,A.J.Mater.Chem.2004,14,1454.doi:10.1039/b400968c
(21) Bard,A.J.;Faulkner,L.R.Electrochemical Methods,2nd ed.;Wiley&Sons:New York,2001;pp 331-332.
(22)Duong,H.T.;Rigsby,M.A.;Zhou,W.P.;Wieckowski,A.J.Phys.Chem.C 2007,111,13460.doi:10.1021/jp072586i
