酸诱导拥挤条件下肌红蛋白及突变体去折叠过程
2013-09-17张玉姣曹洪玉郑学仿
张玉姣 唐 乾 曹洪玉 郑学仿,2,*
(1大连大学生命科学与技术学院,辽宁大连116622;2大连大学辽宁省生物有机化学重点实验室,辽宁大连116622)
1 引言
长期以来,体外模拟反应大都是在稀溶液中进行,而真实的细胞内是高度拥挤的.细胞内含有大量的多糖、蛋白质、核酸等大分子物质,这些大分子以不同浓度存在于细胞内,细胞质内所有大分子的浓度估计高达80-200 g·L-1,1细胞容积的20%-30%都被大分子占用.最近,提出分子伴侣概念的Ellis教授2呼吁生物学家们在研究体系中一定要加入细胞内相应浓度的拥挤试剂以模拟细胞内大分子拥挤环境.Sasahara等3在低pH的拥挤条件下研究脱辅基肌红蛋白构象变化,发现拥挤试剂可以促进去折叠的蛋白质折叠为更为紧致的结构,拥挤试剂的存在增强了蛋白质的稳定性.McPhie等4在拥挤条件下研究脱辅基肌红蛋白的构象变化,发现大分子拥挤试剂浓度的增加能促进去折叠的脱辅基肌红蛋白向熔球态转变,且这种现象与温度无关,但拥挤条件下全辅基肌红蛋白的去折叠是什么过程?是否与脱辅基的肌红蛋白一致?
另外,人们一直认为蛋白表面的氨基酸残基暴露在溶剂中,对蛋白的稳定性影响甚微.但最近几年研究成果对这一观念提出了质疑,Hoffman及其团队5已经发现肌红蛋白三突变体(三个表面的酸性氨基酸Asp44,Asp60,Glu85突变为赖氨酸)显著增强肌红蛋白与细胞色素b5复合体的结合能力及复合体内部电子转移.而且他们也发现马心肌肌红蛋白44位天冬氨酸突变为赖氨酸后,突变体肌红蛋白与细胞色素b5之间的电子传递速率增强了一个级别,6我们课题组已经探讨过肌红蛋白及突变体(D44K,D60K)与各种配体的相互作用,如与表面活性剂7,8以及氧化剂9等,均已证明突变体在与这些配体的作用中有别于野生型蛋白.在拥挤条件下突变体蛋白的去折叠过程是否有别于野生型蛋白呢?因此,本文利用多种光谱学手段研究大分子拥挤条件下全辅基肌红蛋白及突变体D60K去折叠过程,通过比较酸稳定性特点,研究不同环境以及表面氨基酸的改变对蛋白质性质的影响.本项研究可对蛋白质稳定性维持和蛋白质折叠病的研究提供可靠的理论基础.
2 实验部分
2.1 试剂与仪器
马心肌红蛋白样品(美国Sigma公司)UV-Vis吸收光谱特征峰表明几乎全部为高铁肌红蛋白,使用时用Na2HPO4-NaH2PO4缓冲溶液(0.05 mol·L-1)配制成浓度为5.0×10-6mol·L-1的溶液(避光保存并尽快用于实验),突变体Mb(D60K)样品(通过质粒转化后,微生物培养法获得,纯度达96%以上)浓度为5.0×10-6mol·L-1,Ficoll70及Dextran70购于上海生物工程公司,均为国产分析纯.
V-560型UV-Vis分光光度计、FP-6500型荧光分光光度计、J-810型圆二色分光光度计(均购自日本Jasco公司);F-12型制冷和加热循环器(德国Julabo公司).
2.2 实验方法
2.2.1 突变体D60K的获取
细胞培养:将含有Mb(D60K)的质粒转化E.coli BL21感受态细胞,挑取阳性克隆,接种到添加100 mg·L-1的溶菌肉(LB)培养基中大量培养,接种量为1%;离心收集细胞于-18°C冻存备用.
提取:取出冻存的细胞在室温溶解,加入溶菌酶,搅拌,冰浴2-4 h,再加入链霉素并搅拌至细胞提取液为稀溶液.反复清洗细胞碎片,离心收集红色上清液(6000 r·min-1,30 min,4 °C).
纯化:采用硫酸铵分步沉淀.第1步硫酸铵饱和度65%,2 h.第2步硫酸铵饱和度是95%,过夜.离心收集到的沉淀透析除盐,浓缩.浓缩液经过离子交换柱DEAE-Sepharose(1.6 cm×20 cm,pH 7.4,含有1.0×10-3mol·L-1乙二胺四乙酸钠(EDTA)的 Tris-HCl缓冲液).洗脱液再经过Sephadex G-75凝胶柱(1.6 cm×50 cm)纯化.最后得到的纯度达到96%的D60K肌红蛋白,超滤浓缩后-80°C保存.
2.2.2 样品前期处理
将Mb溶解在不同pH值(6.5,6.0,5.5,5.0,4.5,4.0,3.5,3.0)的 0.05 mol·L-1的磷酸盐缓冲液(PBS)中,蛋白终浓度达到5.0×10-6mol·L-1,对照组分别添加Ficoll70与Dextran70,使其终浓度达到100 g·L-1,室温放置1.5 h后进行光谱测定;Mb(D60K)处理同上.
2.2.3 UV-Vis吸收光谱
将已处理的Mb(WT)和Mb(D60K)样品置于1 cm石英比色池中进行测定,测定条件为:狭缝宽度为2 nm,扫描波长范围220-700 nm,扫描速率200 nm·min-1,响应时间中等.
2.2.4 同步荧光光谱
将已处理的Mb(WT)和Mb(D60K)样品置于1 cm石英比色池中进行同步荧光光谱测定,测定条件为:激发狭缝和发射狭缝分别为3和5 nm,扫描速率为500 nm·min-1,响应时间为0.5 s,检测器灵敏度为中等,控制温度(25.00±0.01)°C.
2.2.5 CD光谱
将已处理的Mb(WT)和Mb(D60K)样品置于1 mm石英比色池中进行圆二色谱测定,测定条件为:狭缝宽度为1 nm,扫描波长范围为190-250 nm,扫描速率为50 nm·min-1,响应时间为2 s,累计次数为3次.
3 结果与讨论
3.1 Mb(WT)UV-Vis吸收光谱
Mb(WT)的UV-Vis吸收光谱受血红素周围微环境的影响,天然状态的Mb(WT)溶液在409 nm(Soret)带、503 nm(Q带)、630 nm(金属电子配体转移(LMCT)带)处有特征吸收峰,这分别是血红素的中心铁原子与吡咯环的四个氮原子形成配位键,第五配位结合His-93残基,第六配位结合一个水分子形成的6-cHs(六配位高自旋结构),aquometMb的特征吸收谱带.10图1A为稀溶液中酸诱导Mb(WT)去折叠过程的紫外-可见吸收光谱,在pH 6.5→pH 5.5,Soret带吸光值逐渐增大,pH 5.5→pH 4.0,Soret带的吸光值逐渐下降,当pH为4.0时Soret带的吸光值迅速下降且蓝移至370.5 nm处,该过程中503 nm处的Q带与630 nm处的LMCT带峰位置没有发生改变,当pH为3.5时,503 nm处Q带红移到513 nm,630 nm处的LMCT带红移到644 nm处.Dextran70存在时,pH 6.5→pH 5.5的Soret带变化过程与未加拥挤试剂时相同,但pH降到4.0时,Soret带强度下降幅度较大但峰位置没有改变.当pH降为3.5时,Soret带由409 nm蓝移至370.5 nm处,同时503 nm处的Q带与630 nm处的LMCT带分别红移到512和638.5 nm处,543 nm处出现新的吸收峰.当pH降为3.0时638.5 nm处的吸收峰红移到644 nm.Ficoll70存在时,pH 6.5→pH 3.5,Soret带变化过程与Dextran70存在时相同,但630 nm处LMCT带变化过程与Dextran70存在时不同,pH 3.5时630 nm处吸收峰位置没有发生改变,pH为3.0时630 nm处的吸收峰红移到644 nm.
pH 6.5→pH 5.5血红素周围疏水微环境发生变化,血红素中的二价铁与水相互作用被氧化成高价铁,从而形成了高铁肌红蛋白,使得409 nm处吸收峰值增大.随着pH值继续降低,氨基酸基团质子化造成了血红素结合位点的改变,并使蛋白质构象发生了巨大的改变,11表现为血红素辅基脱离肽链导致409 nm处吸收峰蓝移至370.5 nm处.503 nm处Q带吸收峰红移主要是由于氢离子进入血红素疏水中心,由Gouterman的四轨道模型12和Q带与配体供电子能力的关系可知,溶液供电子能力增加导致肌红蛋白Q带红移.LMCT带由630 nm红移到644 nm,主要是由于氢离子进入血红素活性中心,氢离子与肌红蛋白相互作用增强使得Fe-His-93位的配位键断裂,高铁肌红蛋白(metMb)在血红素活性位点处形成五配位高自旋结构.13Dextran70存在时Soret带蓝移、Q带与LMCT带红移时所处的pH值低于稀溶液,产生此现象的原因是由于Dextran70是无交联的线性大分子,14在溶液中形成准随机螺旋,存在很多可以容纳分子的间隙空间,对Mb(WT)产生不同的排阻体积,降低了血红素与肽链的解离速度,对Fe-His-93位配位键起到了一定的稳定作用,从而对吸收峰转移过程起到了延迟作用.Ficoll70存在时630 nm处LMCT带红移过程迟于Dextran70存在的变性溶液,Ficoll70是高度交联共聚体,产生的排阻体积明显大于Dextran70,阻碍了氢离子进入血红素活性中心,减弱了氢离子与肌红蛋白相互作用,降低了Fe-His-93位的配位键断裂所处的pH值.
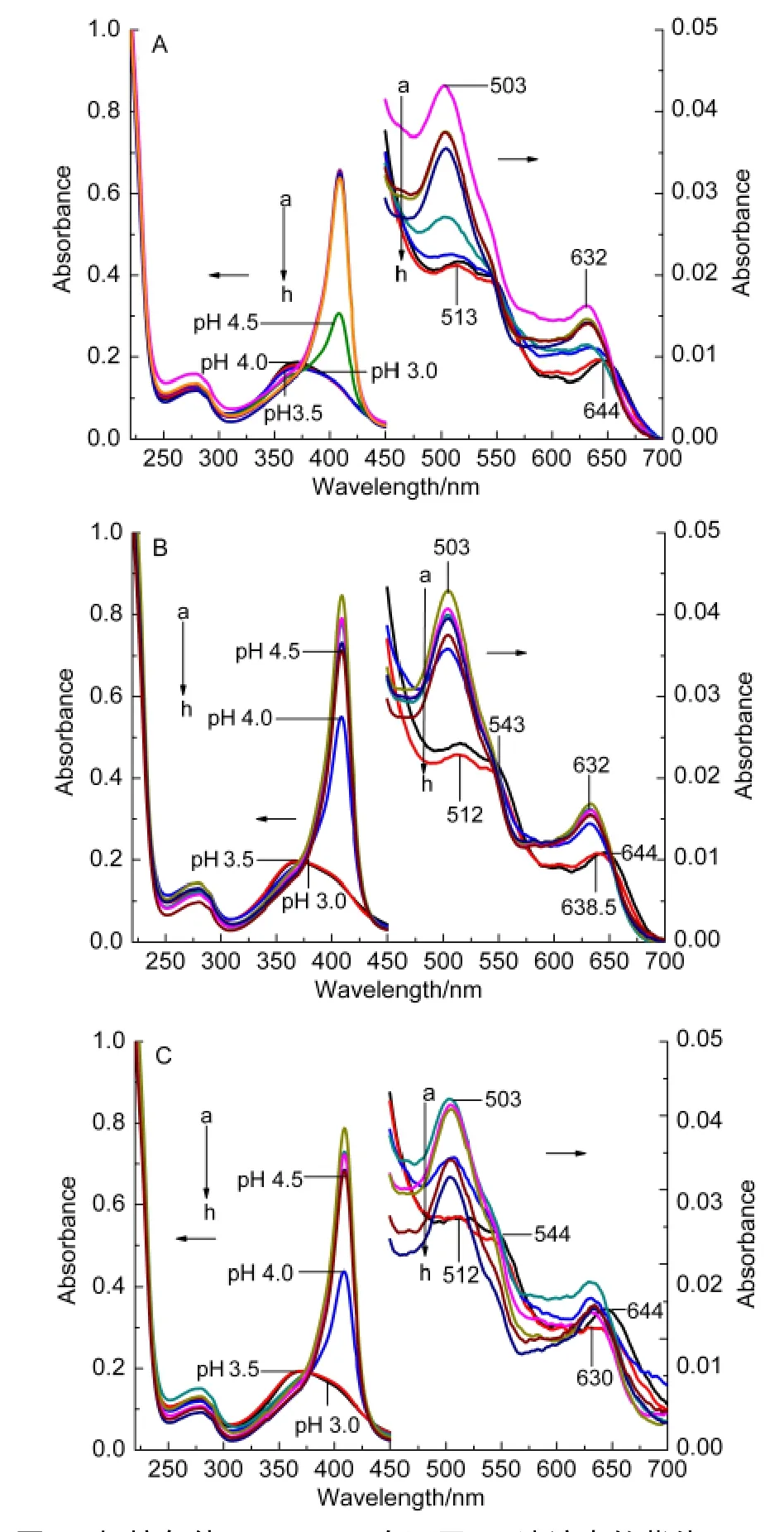
图1 拥挤条件下Mb(WT)在不同pH溶液中的紫外-可见吸收光谱Fig.1 UV-Vis absorbance spectra of Mb(WT)as a function of pH under macromolecular crowding conditions
3.2 Mb(WT)圆二色光谱
通过CD光谱可以灵敏地检测反应引起的蛋白分子的二级结构变化.在蛋白质或多肽的规则二级结构中,肽键是高度有规律排列的,排列的方向决定了肽键能级跃迁的分裂情况,因此具有不同二级结构的蛋白质或多肽所产生的CD谱带的位置、吸收的强弱都不相同.
我们用远紫外区圆二色光谱技术检测Mb(WT)的二级结构变化情况,并用杨氏方程算法15计算Mb(WT)中的α螺旋、β折叠等含量变化.208和222 nm处两个负槽,反映的是α螺旋的光谱特性.16图2为酸诱导Mb(WT)去折叠过程的圆二色光谱,稀溶液中pH 6.5→pH 4.5,Mb(WT)208和222 nm处的负槽强度下降但形状和肩峰的位置没有发生明显的改变.蛋白质在此pH值范围内没有变性,整个蛋白分子结构仍以α螺旋为主,在pH 4.5时无拥挤试剂的Mb(WT)二级结构有较大改变,α螺旋含量下降13.9%,这主要是由于大量的氢离子作用于肌红蛋白表面使蛋白伸展并暴露出疏水残基干扰蛋白的螺旋结构,更多的β折叠及无规则卷曲可溶性暴露结构出现.随着酸度的增强蛋白质表面的正电荷增强使得位于蛋白表面的亲水性侧链基团发生了明显的变化,维系二级结构的氢键作用力被破坏,对酸敏感的二级结构(α-螺旋及伸展肽链)开始解离,17β折叠、无规则卷曲含量在增加,蛋白质的有序度在降低,无序度在增加.18与稀溶液相比,拥挤试剂Dextran70与Ficoll70存在的蛋白溶液的负槽强度减弱程度低很多,但其二级结构仍遭到破坏,在pH 4.5时α螺旋含量分别下降9.5%、4.5%,此时拥挤试剂形成了高度拥挤的状态降低了氢离子的扩散系数,阻碍了氢离子的运动,减弱了氢离子与蛋白质之间的相互作用力,蛋白质的二级结构改变值明显低于无拥挤试剂的蛋白质溶液,拥挤试剂的加入对肌红蛋白二级结构起到了保护作用.pH降到4.0时稀溶液中蛋白在208和222 nm处两个负槽的形状与强度发生了明显的改变,而Dextran70与Ficoll70存在的溶液基本没有发生改变,pH降到3.5时拥挤试剂存在的蛋白溶液的负槽形状与强度发生明显改变,蛋白质二级结构遭到严重破坏,CD光谱检测的蛋白质二级结构变化过程与紫外-可见光谱检测的蛋白质空间构象的变化过程相一致.
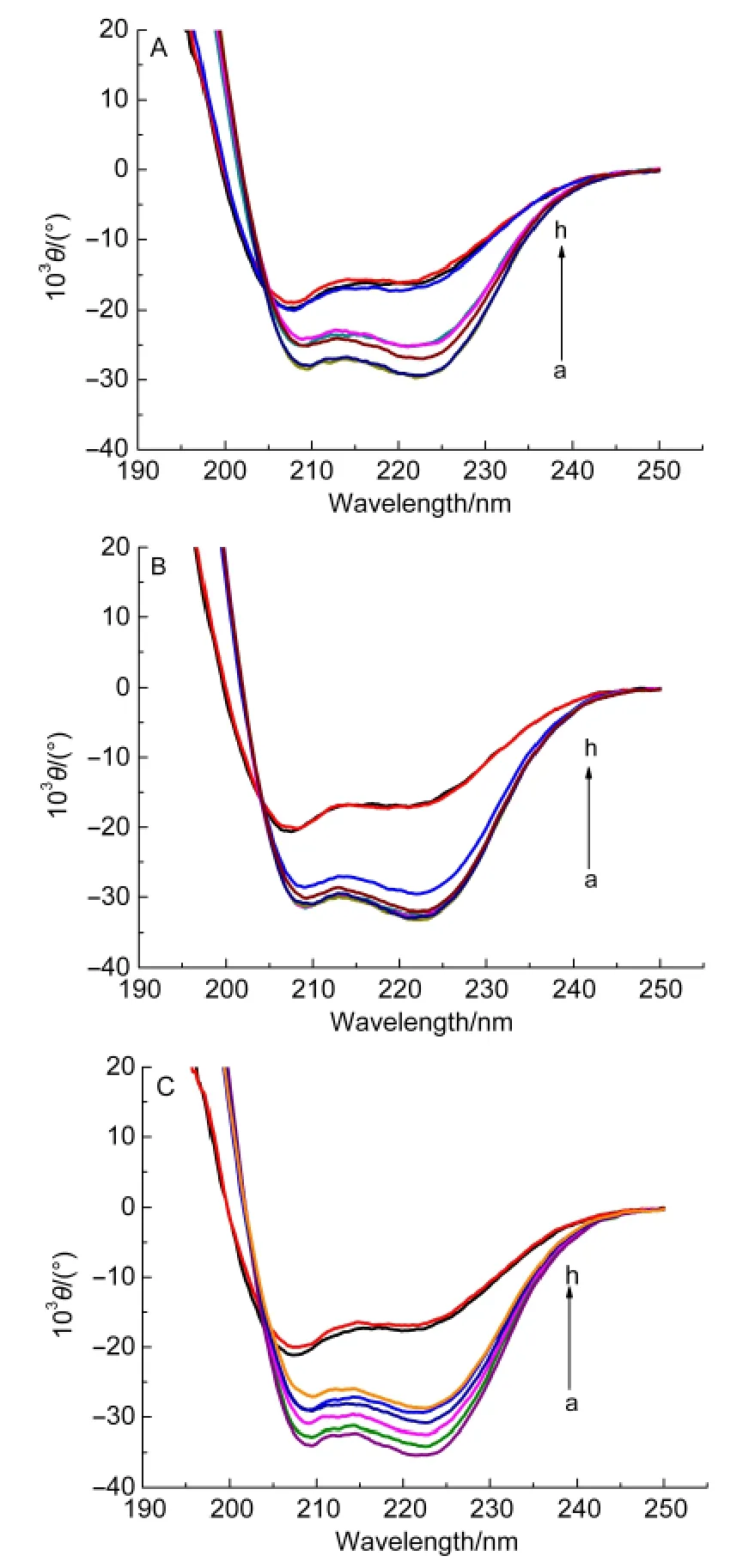
图2 拥挤条件下Mb(WT)在不同pH溶液的圆二色光谱Fig.2 CD spectra of Mb(WT)as a function of pH under macromolecular crowding conditions
3.3 Mb(WT)紫外数据归一化处理
图3为拥挤条件下Mb(WT)在不同pH溶液中409 nm处吸收值的变化图谱,通过紫外光谱与CD光谱可以清楚地看到稀溶液与拥挤条件下Mb(WT)血红素辅基以及肽链的变化情况,为了更精确地比较拥挤试剂加入前后蛋白去折叠过程的差异,依据文献19计算出变性中点pH(pH1/2),计算结果显示稀溶液中Mb(WT)酸变性中点pH1/2(Mb)=4.25,Dextran70存在时pH1/2(Mb)=3.78,Ficoll70存在时pH1/2(Mb)=3.76.由此可见,拥挤试剂加入后Mb(WT)酸变性中点pH明显下降,Mb(WT)的耐酸性提高,拥挤试剂具有酸变性过程中稳定肌红蛋白结构的作用.
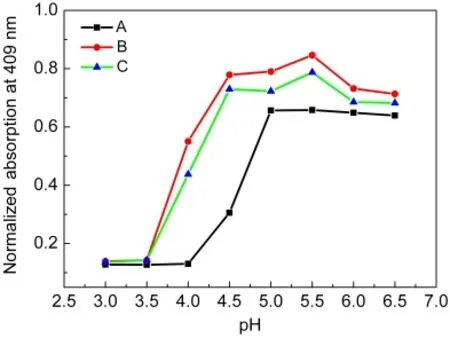
图3 拥挤条件下Mb(WT)在不同pH溶液中409 nm处归一化吸收值的变化Fig.3 Normalized absorption values of Mb(WT)at 409 nm as a function of pH under macromolecular crowding conditions
3.4 Mb(WT)荧光相图分析
荧光相图法是根据内源荧光发射光谱特定部位的荧光强度作图,进而直观地获得蛋白质去折叠结构变化信息的方法.20发射波长分别为λ1和λ2的荧光强度分别为I(λ1)和I(λ2).变量间存在以下三个关系式:21-23

式中a和b分别为I(λ1)对I(λ2)作图所得直线的截距和斜率,I1(λ1)和 I2(λ1)分别为λ1条件下蛋白质结构发生变化时始态和终态的荧光强度,I1(λ2)和I2(λ2)则分别是发射波长为λ2条件下蛋白质结构发生变化时始态和终态的荧光强度.分别选取波长为320和365 nm处的荧光强度为研究对象.
若蛋白质去折叠过程符合“全或无模型”或“二态模型”,荧光相图表现为一条直线;若符合“三态”或“多态”模型,荧光相图分别表现为两条或多条直线.24-26
图4A为稀溶液中酸诱导Mb(WT)去折叠过程荧光相图,该荧光相图表现为一条直线,表明稀溶液中随着酸度增强Mb(WT)发生去折叠且去折叠过程为“二态”模型.图4B、图4C为拥挤试剂加入后Mb(WT)去折叠过程荧光相图,该荧光相图仍旧为一条直线,拥挤试剂的加入没有增加Mb(WT)去折叠过程的中间态,图中各pH值所对应的荧光相图结果与紫外光谱、CD光谱变化结果相一致.
通过光谱数据与实验计算结果可以看出:拥挤试剂的加入对Mb(WT)的空间构象以及氨基酸的微环境起到了很好的保护作用,而突变体Mb(D60K)的酸变性过程又是怎样呢?
3.5 Mb(D60K)UV-Vis吸收光谱
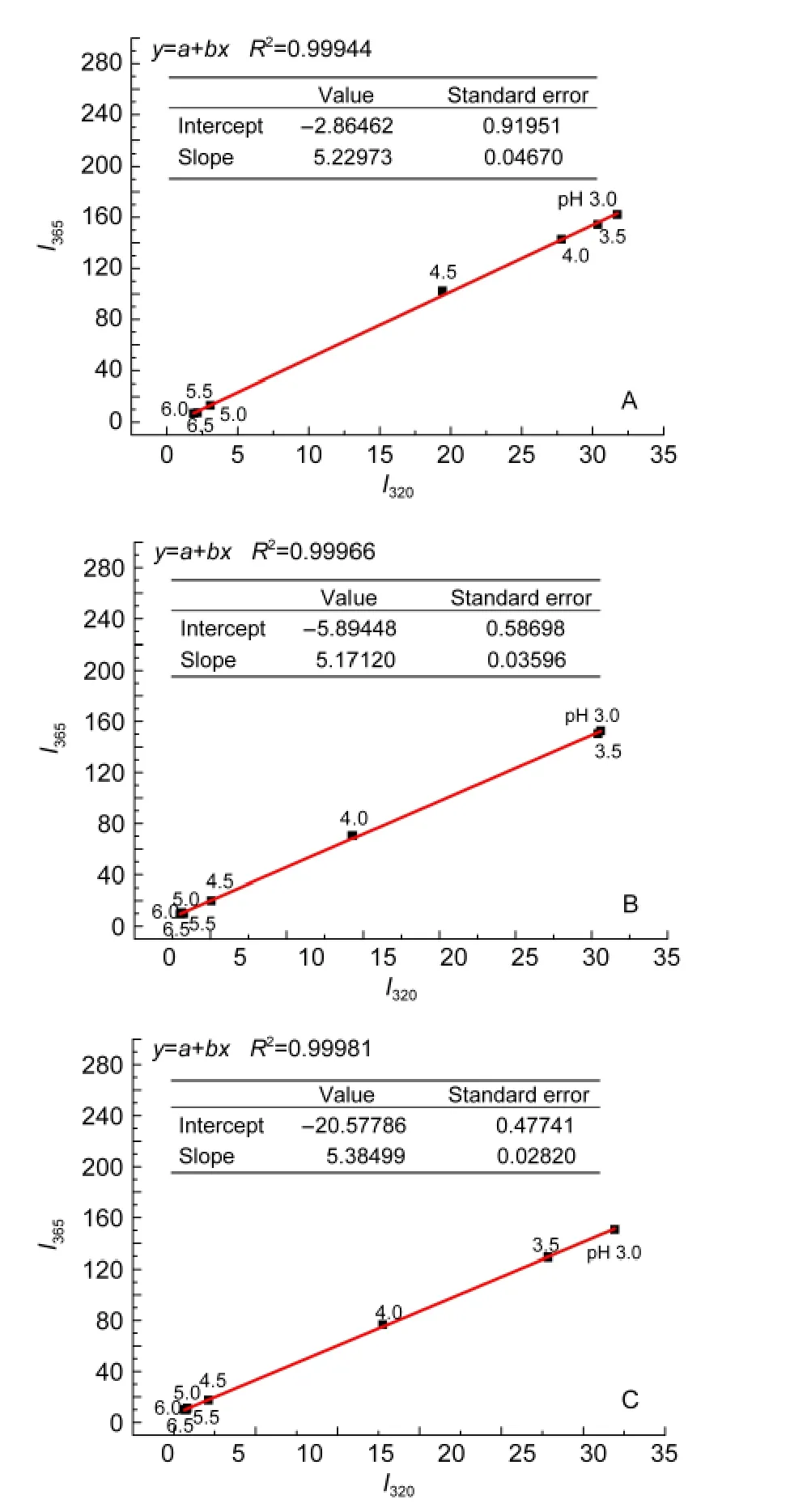
图4 拥挤条件下Mb(WT)在不同pH溶液中的荧光相图Fig.4 Fluorescence phase diagram of Mb(WT)as a function of pH under macromolecular crowding conditions
图5A为稀溶液中酸诱导Mb(D60K)去折叠过程紫外-可见吸收光谱,肌红蛋白60位Asp突变为Lys后其去折叠过程与野生型有一定的区别,D60K约在542、580 nm处有特征吸收峰,此处是还原型蛋白的特征吸收峰,pH 6.5→pH 4.5,Soret带的变化过程与野生型相同,pH为4.0时Mb(D60K)409 nm处吸收峰蓝移至387 nm处(Mb(WT)则蓝移至370.5 nm)且503、542以及约580 nm处吸收峰消失,630 nm处吸收峰红移到644 nm处,当pH为3.5时409 nm处最大吸收峰蓝移至370.5 nm.上述现象产生的原因主要是随着酸度增强,溶液的pH值小于赖氨酸的等电点时,Mb(WT)的Lys-60上―NH2全都变为―NH3+,蛋白表面整体偏正电荷,而溶液pH值大于天冬氨酸的等电点时,Asp-60上的―COOH全都变为―COO-,蛋白表面偏负电荷;Mb的60位Asp突变为Lys改变了蛋白质表面的电荷,弱化了蛋白质表面电荷受pH值的影响,蛋白质与氢离子之间的相互作用减弱,从而延迟了最大吸收峰移动.
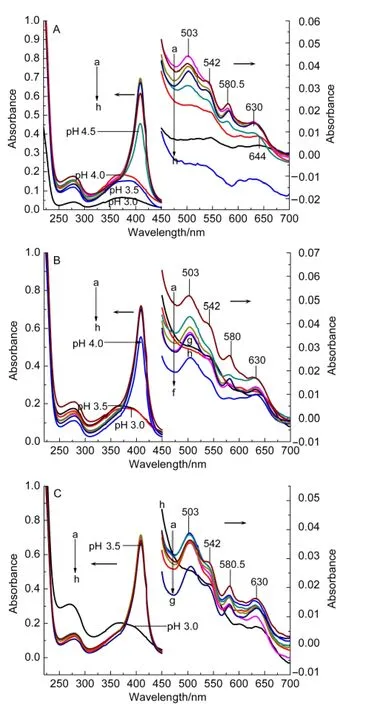
图5 拥挤条件下Mb(D60K)在不同pH溶液中的UV-Vis吸收光谱Fig.5 UV-Vis absorbance spectra of Mb(D60K)as a function of pH under macromolecular crowding conditions

图6 拥挤条件下Mb(D60K)在不同pH溶液的圆二色光谱Fig.6 CD spectra of Mb(D60K)as a function of pH under macromolecular crowding conditions
Dextran70存在时,pH 6.5→pH 4.5,各吸收峰变化过程与稀溶液相同,pH为4.0时,409 nm处最大吸收峰强度稍有下降但位置没有发生改变,542、580 nm处吸收峰消失但503 nm处Q带吸收峰与630 nm处的电子转移带没有发生改变(稀溶液中409 nm处最大吸收峰蓝移至387 nm)且503、542 nm以及580 nm处吸收峰消失,630 nm处吸收峰红移到644 nm处.当pH降为3.5时,最大吸收峰蓝移至373.5 nm处且503 nm处的吸收峰消失,蓝移程度都明显低于无拥挤试剂存在的Mb(D60K)溶液,pH降为3.0时最大吸收峰蓝移至370.5 nm,此时蛋白质已变性,血红素辅基完全脱离肽链.拥挤试剂Dextran70形成高度拥挤的环境降低了氢离子的扩散系数,减弱了氢离子与蛋白质的相互作用,从而延迟了蛋白质的变性过程.
Ficoll70存在时,pH 6.5→pH 3.5各吸收峰强度及峰位置基本没有变化,当pH降到3.0时,409 nm处吸收峰强度迅速下降且蓝移至370.5 nm处,503 nm处Q带吸收峰以及约542、580 nm处吸收峰消失,但630 nm处的LMCT带未发生改变,中间没有经历任何中间态,说明拥挤试剂Ficoll70的加入增强了突变体Mb(D60K)的酸稳定性且拥挤试剂Ficoll70对Mb(D60K)的稳定效果明显强于Dextran70.
3.6 Mb(D60K)圆二色光谱
图6为拥挤条件下Mb(D60K)在不同pH溶液的圆二色光谱,稀溶液中pH 6.5→pH 4.5,D60K酸变性过程与Mb(WT)相同,但pH 4.5时α螺旋含量下降6.2%,改变值明显低于Mb(WT);拥挤试剂Dextran70存在时pH 6.5→pH 4.0,208和222 nm处的负槽强度基本没有变化,但在pH 4.0时其α螺旋含量下降5.8%;Ficoll70存在时pH 6.5→pH 3.5,208和222 nm处的负槽强度基本没有变化,只有在pH 3.0时负槽强度发生明显变化,此时α螺旋含量下降7.4%.表面氨基酸突变与拥挤试剂的加入均增强了Mb(D60K)二级结构稳定性.

图7 拥挤条件下Mb(D60K)在不同pH溶液中409 nm处吸收值的变化Fig.7 Normalized absorption value of Mb(D60K)at 409 nm as a function of pH under macromolecular crowding conditions
3.7 Mb(D60K)紫外数据归一化处理
图7为拥挤条件下Mb(D60K)在不同pH溶液中409 nm处吸收值的变化图谱,通过谱图数据计算D60K变性的中点pH,稀溶液中pH1/2(D60K)=4.19,Dextran70存在时pH1/2(D60K)=3.74,Ficoll70存在时pH1/2(D60K)=3.12.从上述数据可以看出肌红蛋白第60位天冬氨酸突变为赖氨酸后酸变性中点降低,Dextran70存在时Mb(D60K)的变性中点pH值降低10.74%,Ficoll70存在时变性中点pH值降低了25.72%,拥挤试剂具有降低蛋白质酸变性中点的作用.
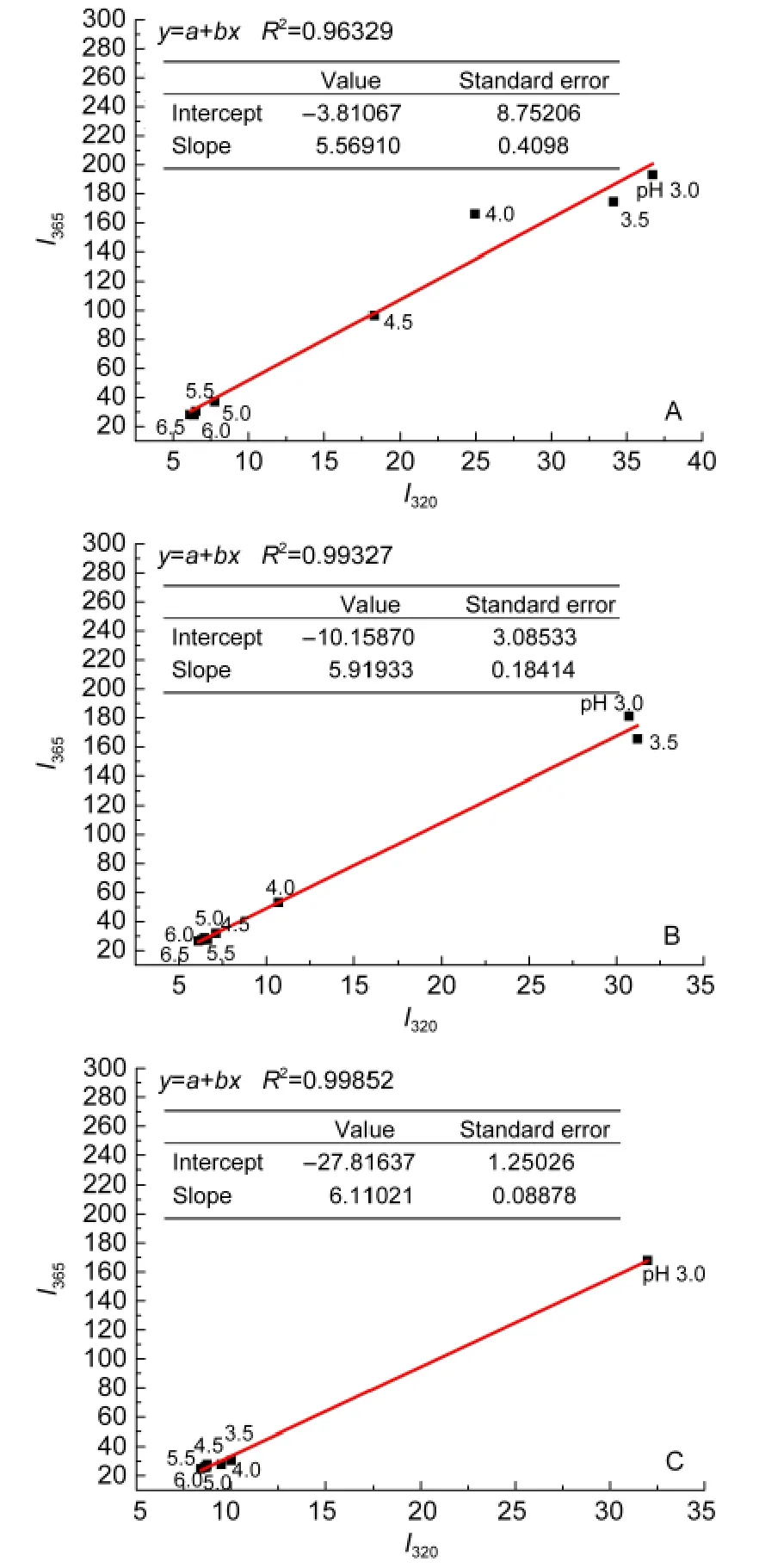
图8 拥挤条件下Mb(D60K)在不同pH溶液的荧光相图Fig.8 Fluorescence phase diagram of Mb(D60K)as a function of pH under macromolecular crowding conditions
3.8 酸诱导Mb(D60K)去折叠过程荧光相图分析
图8为拥挤条件下Mb(D60K)在不同pH溶液的荧光相图,从图中可以看出添加了拥挤试剂后Mb(D60K)去折叠过程仍为二态过程,拥挤试剂的加入并未增加Mb(D60K)去折叠中间态,拥挤试剂对Mb(D60K)的去折叠中间态没有影响.
4 结论
本文通过加入大分子拥挤试剂,模拟高度拥挤的细胞内环境,研究酸诱导Mb(WT)及其突变体Mb(D60K)去折叠过程,结果显示肌红蛋白表面60位天冬氨酸突变为赖氨酸后增强了肌红蛋白结构稳定性;大分子拥挤试剂加入后Mb(WT)及其突变体的去折叠过程明显不同于稀溶液,拥挤试剂提高了Mb(WT)及Mb(D60K)的酸变性中点且Ficoll70对Mb(D60K)耐酸性的提高效果尤为显著.肌红蛋白表面氨基酸突变以及拥挤试剂的添加起到了稳定血红素微环境、芳香族氨基酸、蛋白质二级结构和保护蛋白天然状态的作用,为以后拥挤条件下蛋白质去折叠过程的研究提供参考依据.
Supporting Information: The original data of fluorescence emission spectrum are used to calculate the fluorescence phase diagram of Mb(WT)induced by acid.This information is available free of charge via the internet at http://www.whxb.pku.edu.cn.
(1)Swaminathan,R.;Hoang,C.P.;Verkman,A.S.Biophys.J.1997,72(4),1900.doi:10.1016/S0006-3495(97)78835-0
(2) Ellis,R.J.Curr.Opin.Struct.Biol.2001,11(1),114.doi:10.1016/S0959-440X(00)00172-X
(3) Sasahara,K.;McPhie,P.;Minton,A.P.J.Mol.Biol.2003,326(4),1227.doi:10.1016/S0022-2836(02)01443-2
(4) McPhie,P.;Ni,Y.S.;Minton,A.P.J.Mol.Biol.2006,361(1),7.doi:10.1016/j.jmb.2006.05.075
(5) Hoffman,B.M.;Celis,L.M.;Cull,D.A.;Patel,A.D.;Seifert,J.L.;Wheeler,K.E.;Wang,J.;Yao,J.;Kurnikov,I.V.;Nocek,J.M.Proc.Natl.Acad.Sci.U.S.A.2005,102(10),3564.doi:10.1073/pnas.0408767102
(6)Ma,J.Y.;Ma,J.;Zheng,X.F.;Tang,Q.;Gao,D.B.Chin.J.Anal.Chem.2008,36(4),454.[马君燕,马 静,郑学仿,唐 乾,高大彬.分析化学,2008,36(4),454.]
(7)Li,Y.W.;Cao,H.Y.;Tang,Q.;Zheng,X.F.Acta Phys.-Chim.Sin.2010,26(6),1687. [李宜雯,曹洪玉,唐 乾,郑学仿.物理化学学报,2010,26(6),1687.]doi:10.3866/PKU.WHXB20100617
(8)Zhang,Y.Y.;Cao,H.Y.;Tang,Q.;Zheng,X.F.Acta Phys.-Chim.Sin.2011,27(12),2907.[张莹莹,曹洪玉,唐 乾,郑学仿.物理化学学报,2011,27(12),2907.]doi:10.3866/PKU.WHXB20112907
(9)Zhi,Q.Y.;Tang,Q.;Cao,H.Y.;Zhang,Y.Y.;An,L.M.;Zheng,X.F.Spectroscopy and Spectral Analysis 2011,31(9),2512.[职秋艳,唐 乾,曹洪玉,安良梅,张莹莹,郑学仿.光谱学与光谱分析,2011,31(9),2512.]
(10) Eaton,W.A.;Hochstrasser,R.M.J.Chem.Phys.1968,49(3),985.doi:10.1063/1.1670263
(11) Culbertson,D.S.;John,S.L.Biochemstry 2010,49(29),6052.doi:10.1021/bi1006942
(12)Zerner,M.;Gouterman,M.;Kobayashi,H.Theor.Chim.Acta 1996,6,363.
(13) Dua,J.;Sonoa,M.;Dawson,J.H.J.Porphyrins Phthalocyanines 2011,15,29.doi:10.1142/S1088424610002872
(14) Zhao,M.;Jing,J.;Li,S.Journal of Beijing Normal University 2007,43(4),442.[赵 明,井 健,李 森.北京师范大学学报,2007,43(4),442.]
(15) Chen,Y.H.;Yang,J.T.;Chau,K.H.Biochemistry 1974,13(33),3335.
(16)Taheri-Kafrani,A.;Asgari-Mobarakeh,E.;Bordbar,A.K.;Haertle,T.Colloids Surf B-Biointerfaces 2010,75(1),268.doi:10.1016/j.colsurfb.2009.08.045
(17)Wang,Q.;Run,Y.B.;Sun,S.Q.;Zhou,Q.;Hu,X.R.Journal of Guang Xi Normal University 2003,21(2),255.[王 琪,闰永彬,孙素芹,周 群,胡鑫尧.广西师范大学学报,2003,21(2),255.]
(18)Wu,Z.J.;Kang,L.L.;Huang,B.T.;Wu,Y.;Luo,M.;Huang,Y.X.Acta Biophysica Sinica 2009,25(S1),334.[吴正洁,康立丽,黄宝添,吴 越,罗 曼,黄耀熊.生物物理学报,2009,25(S1),334.]
(19) Shosheva,A.;Miteva,M.;Christova,P.;Atanasov,B.Biophys.J.2003,31(8),617.
(20) Kuznetova,I.M.;Turoverov,K.K.;Uversky,V.N.Journal of Proteome Research 2004,3(3),485.doi:10.1021/pr034094y
(21) Bushmarina,N.A.;Kuznetsova,I.M.;Biktashev,A.G.;Turoverov,K.K.;Uversky,V.N.ChemBioChem 2001,2(11),813.doi:10.1002/1439-7633(20011105)2:11<813::AID-CBIC813>3.0.CO;2-W
(22)Kuznetsova,I.M.;Stepanenko,O.V.;Turoverov,K.K.;Zhu,L.;Zhou,J.M.;Fink,A.L.;Uversky,V.N.Biochim.Biophys.Acta 2002,1596(1),138.doi:10.1016/S0167-4838(02)00212-1(23)Turoverov,K.K.;Verkhusha,V.V.;Shavlovsky,M.M.;Biktashev,A.G.;Povarova,O.I.;Kuznetsova,I.M.Biochemistry 2002,41(3),1014.doi:10.1021/bi015548c
(24) Chang,J.Y.;Li,L.FEBS Letters 2002,511(1-3),73.doi:10.1016/S0014-5793(01)03284-7
(25) Sasahara,K.;Demura,M.;Nitta,K.Proteins 2002,49(4),472.
(26) Laurents,D.;Baldwin,R.Biochemistry 1997,36(6),1496.doi:10.1021/bi962198z
