纳米三氧化钨复合催化剂的制备及对甲醇电催化性能
2013-09-17刘委明胡仙超褚有群马淳安
周 阳 刘委明 胡仙超,3 褚有群 马淳安,*
(1浙江工业大学化工材料学院,绿色化学合成技术国家重点实验室培育基地,科技部能源材料及应用国际科技合作基地,杭州310032;2江西理工大学冶金与化学工程学院,江西赣州341000;3浙江工业大学分析测试中心,杭州310032)
1 Introduction
Direct methanol fuel cells(DMFCs)have recently attracted considerable attentions due to their excellent features such as high-energy density,convenient fuel storage,green emission,and ambient operating conditions etc.1,2However,DMFCs usually use lots of expensive platinum as anodic catalysts that tend to be poisoned by reaction intermediates such as COads.3Thus considerable efforts have been devoted to making metallic alloy,such as PtRu,4,5PtPd,6,7and PtRuSn8,9etc.,with low amount of platinum and high activity toward methanol oxidation.But the dissolution of transition metals in the alloys during the DMFC operation would be the main challenge since the dissolved transition metals may span the membrane and experience reduction on the cathode,finally leading to the unexpected performance degradation of DMFCs.10,11
The second way to design Pt-based composite catalysts is metal oxides modified Pt particles,such as Pt/RuO2,12,13Pt/SnO2,14,15and Pt/MnO2,16etc.Tungsten trioxide(WO3)is known to be able to form a hydrogen tungsten bronze(HxWO3)compound in acid solution which is both nonstoichiometric and electrically conducting.The compound can facilitate dehydrogenation during methanol oxidation and lighten the CO poisoning of Pt catalyst.Previous studies have shown that Pt and PtRu catalysts supported on WO3have extremely high activity towards the electro-oxidation of CO,17methanol,18-21ethanol,22and formic acid.23,24However,WO3has a low specific surface area and conductivity,which limits its application in DMFC.
Recently WO3/C hybrid material was used as the support of Pt-WO3/C catalysts.25,26Compared with Vulcan XC-72 carbon black,carbon nanotubes(CNTs)have better specific surface area and conductivity.Rajesh et al.27reported a composite catalyst of methanol electro-oxidation by depositing Pt nanoparticles on WO3-modified CNT,in which CNT was synthesised by the template carbonisation of polypyrrole on alumina membrane.In this paper CNT was further disposed by strong acid so that Pt and WO3nanoparticles were homogenously deposited on the surface of CNT.Results show that WO3-modification improves significantly electrocatalytic activity towards methanol oxidation.
2 Experimental
2.1 Preparation of WO3modified acid treated CNTs
Carbon nanotubes(Shenzhen Nanoharbor Co.,China)were functionalized in nitric acid(65%-68%)under refluxing at 150°C for 5 h,washed by distilled water and dried in vacuum at 85°C.28WO3-modified carbon nanotubes(WO3-CNTs)were prepared by the conventional means with sodium tungstate as the precursor.Briefly,50 mg of CNTs was added into 5 mmol·L-1aqueous solution of sodium tungstate.After ultrasonic dispersed for 30min,the solution were stirred vigorously at 60°C for 1 h,then excessive 1 mol·L-1hydrochloric acid was dropwised into the above solution.After the reaction proceeded for 6 h,the suspension was filtered,washed and dried at 80°C in a vacuum oven.The resultant was transferred into a tubular oven and heat-treated at 500°C for 6 h under the protection of a nitrogen atmosphere.The ideal ratio of WO3to CNTs was calculated as 25%(w),but for comparison,ratio of WO3to CNTs with 10%,25%,50%,and 75%(w)were also prepared following same procedures as above.
2.2 Synthesis of Pt nanoparticles on WO3-CNTs
Platinum supported on the WO3-modified CNTs(Pt/WO3-CNTs)was prepared by means of microwave heating ethylene glycol method.In brief,5.7 mL of 5 mmol·L-1chloroplatinic acid was well mixed with 15 mL ethylene glycol(EG)in a special reaction tube,and then 50.0 mg of as-prepared WO3/CNTs was added into the mixture.After the pH of the mixture was adjusted to 10 using 1.0 mol·L-1NaOH aqueous,well-dispersed slurry was obtained after being stirred in an ultrasonic bath for 30 min.Thereafter,the slurry was microwave-heated at 160°C for 30 min in the microwave synthesizer(Initiator Biotage,Sweden).The resulting solution was filtered,washed and dried at 85°C for 10 h in a vacuum oven,yielding 10%(w)Pt loading on the supports.As contrast samples,Pt nanoparticles(10%(w)metal content)on acid treated CNTs(Pt/CNTs)was prepared using similar procedures as described above.
2.3 Characterizations
The morphology,crystal phase,structure and element distribution of the samples were respectively characterized by XRD,XPS,and TEM.XRD was performed with a Thermo ARL SCINTAG XʹTRA X-ray at room temperature,using quartz monochromatic Cu Kα1radiation source(λ=0.1541 nm)under a voltage of 45 kV and a current of 40 mA.The XRD patterns were recorded with a step size of 0.04°from 10°to 80°at the speed of 2.4(°)·min-1.TEM was carried out on a Tecnai G2 F30 S-Twin(Philips-FEI).XPS was carried out on Kratos AXIS Ultra DLD.
2.4 Electrochemical measurements
Electrochemical measurements were performed on Ivium electrochemical workstation.A standard three-electrode cell with separate anode and cathode compartments was used.A Pt foil and saturated calomel electrode(SCE)were used as counter and reference electrodes,respectively.For electrode preparation,2.5 mg of electrocatalyst sample was ultrasonically mixed in 400 μL of ethanol-water solution(1:1,V/V)to form a homogeneous ink followed by dropping 5 μL of the electrocatalyst ink onto the surface of a glassy carbon electrode(GCE,with a diameter of 3 mm),and 7 μL of Nafion solution of 1.0%(DuPont,USA)in ethanol was added to fix the electrocatalyst on the GCE surface.The electrochemical active surface(EAS)assessed in a nitrogen-saturated 0.5 mol·L-1H2SO4solution at a scan rate of 50 mV·s-1and the electrocatalytic activity for the methanol oxidation reaction was measured in a nitrogensaturated 0.5 mol·L-1H2SO4+1.0 mol·L-1CH3OH solution at a scan rate of 50 mV·s-1.
The CO stripping experiments were performed in a 0.5 mol·L-1H2SO4solution.Along with the continuous CO bubbling for 30 min,the anode electrode was controlled at-0.14 V for CO adsorption.The solution was then purged with N2for 30 min to remove the dissolved CO before the stripping test.
3 Results and discussion
3.1 XRD and TEM analysis of samples
Fig.1 shows the typical XRD patterns of the samples.The diffraction peak at 2θ=26.2°is characteristic of the graphite(002)plane,demonstrating the graphitization of carbon in the sample.The distinct diffraction peaks at 2θ of 23.09°,23.58°,24.33°,33.25°,34.12°,and 41.44°are indexed as the(002),(020),(200),(022),(202),and(222)planes of monoclinic WO3phase.29Those slight diffraction peaks at 2θ of 39.76°,46.28°,and 67.53°are attributed to the Pt(111),(200),and(220)planes,which are not obvious for Pt/WO3-CNTs,indeed,the broaden peak centered at 41.45°is the overlapped peak of the(111)peak of Pt and the(222)peak of WO3.
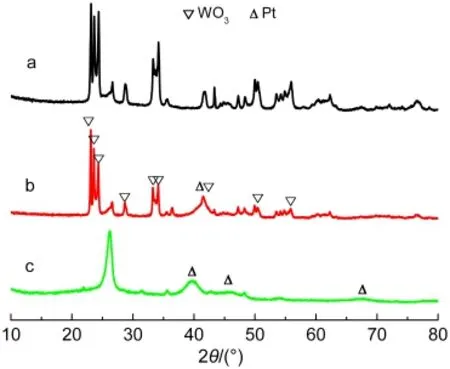
Fig.1 XRD patterns of(a)WO3-CNTs,(b)Pt/WO3-CNTs,and(c)Pt/CNTs catalysts
Fig.2 shows scanning transmission electron microscope(STEM)images of Pt/CNTs and Pt/WO3-CNTs catalyst.The black and white picture is sample particle morphology and color pictures are samplesʹelements distribution density by energy dispersive spectrometer(EDS)surface scan.As can be seen from Fig.2(a),W,Pt,and O elements are distributed on the outer surface of CNTs.Combined with XRD characterization of results,Pt/WO3-CNTs catalyst is composed of Pt,WO3,and CNTs.
Fig.3 gives the TEM images of Pt/CNTs and Pt/WO3-CNTs catalysts and the corresponding histograms of the Pt particle diameters,as well.It can be seen from the TEM images that the Pt particles on the WO3-CNTs support are smaller and more uniformly dispersed than those on CNTs support.The average sizes of the Pt particles in Pt/CNTs and Pt/WO3-CNTs catalysts are estimated from their histograms as being approximately 4.8 and 3.6 nm,respectively,indicating that the introduction of WO3can inhibit the aggregation of Pt particles.26
3.2 XPS analysis of samples
Fig.4 shows the XPS spectra of Pt 4f and W 4f photoemission from Pt/WO3-CNTs,respectively.The two characteristic peaks observed in the Pt 4f region with binding energies of 71.5 and 74.8 eV should be attributed to the metallic Pt.30The two peaks in the W 4f region with binding energies centered at 35.7 and 37.9 eV,suggest the presence of tungsten in the+VI oxidation state.31Fig.5 displays the XPS spectra of Pt 4f photoemission from Pt/CNTs.It can be observed that the two peaks with binding energies of 71.5 and 74.8 eV are characteristic of the metallic Pt,and the other two peaks at 72.3 and 75.8 eV can be assigned to Pt2+in PtO and Pt(OH)2-like species.32
3.3 Electro-catalytic performance

Fig.2 EDS elemental mapping of(a)Pt/WO3-CNTs and(b)Pt/CNTs catalysts under STEM model
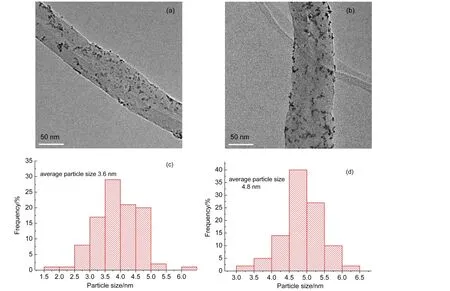
Fig.3 TEM images of(a)Pt/WO3-CNTs and(b)Pt/CNTs,and Pt particle size distributions of(c)Pt/WO3-CNTs and(d)Pt/CNTs
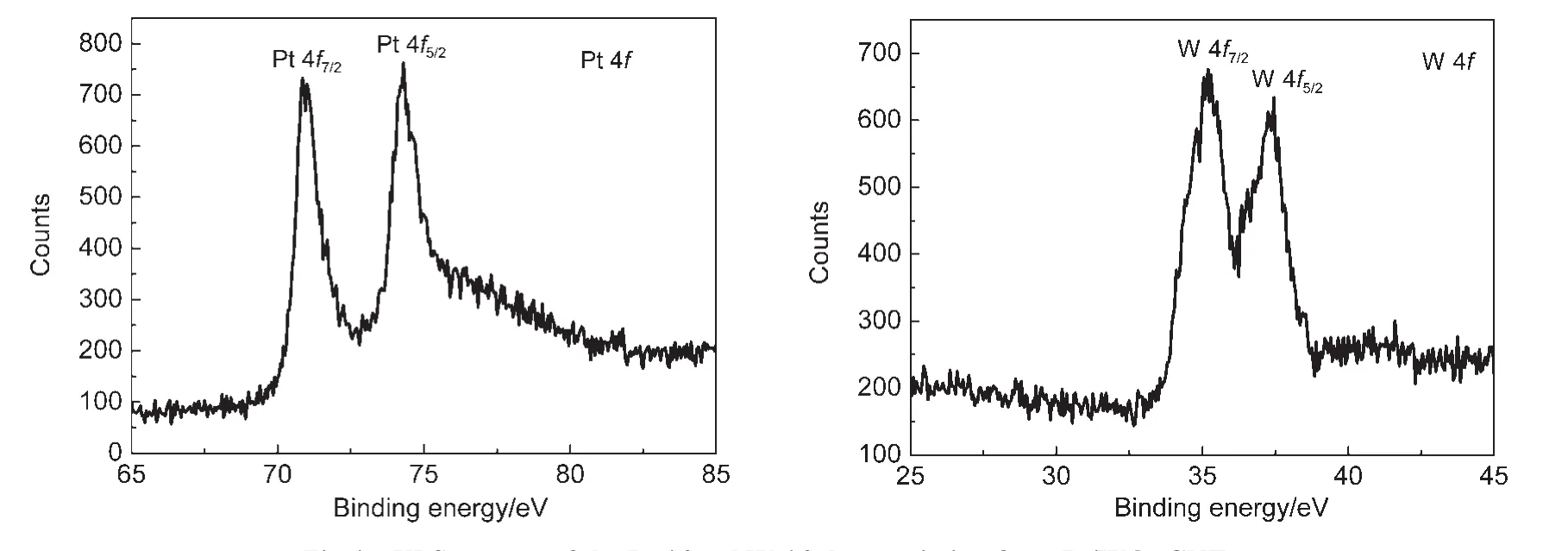
Fig.4 XPS spectra of the Pt 4f and W 4f photoemission from Pt/WO3-CNTs
Fig.6 presents cyclic voltammograms(CVs)of Pt/CNTs and Pt/WO3-CNTs in 0.5 mol·L-1H2SO4solution at a scan rate of 50 mV·s-1.The platinum in Pt/WO3-CNT has a larger EAS than that in Pt/CNTs,which is reflected by the hydrogen adsorption/desorption currents at the potentials between-0.2 and 0.2 V,as shown in Fig.6.The EAS of platinum can be calculated from the integrated charge in the hydrogen adsorption region of the cyclic voltammograms(Fig.6)based on ESA=QH/0.21×[Pt],where QHis the integrated charge(mC),[Pt]is the Pt loading(mg·cm-2)on the electrode.The EAS values calculated for Pt/CNTs and Pt/WO3-CNTs are shown in Table 1,which reveals that the EAS of platinum is influenced by the particle sizes.This result is consistent with Pt particle size distributions of Pt/CNTs and Pt/WO3-CNTs(Fig.3).
Fig.7 shows CVs of the electrodes in 0.5 mol·L-1H2SO4+1 mol·L-1CH3OH solution between 0.0 to 1.0 V at a scan rate of 50 mV·s-1.The electrocatalytic activity of Pt/WO3-CNTs and Pt/CNTs catalysts on the oxidation of methanol was studied in 0.5 mol·L-1H2SO4aqueous solution containing 1.0 mol·L-1CH3OH at a scan rate of 50 mV·s-1.It can be observed from Fig.7 that the onset of methanol oxidation peaks for the Pt/WO3-CNTs catalyst is at 0.25 V,which is apparently lower than that on Pt/CNTs catalysts with the onsets at 0.30 V.The negative shift on the potential onset of Pt/WO3-CNTs indicates that Pt nanoparticles on WO3-CNTs surface can effectively reduce the over potentials in the methanol electro-oxidation reaction.In addition,it can be seen that the mass specific current of Pt/WO3-CNTs(403 mA·mg-1)is 5 times that of Pt/CNT(80 mA·mg-1)at 0.69 V,indicating that WO3plays a key role in the high catalytic performance.

Fig.5 XPS spectra of the Pt 4f photoemission from Pt/CNTs

Fig.6 Cyclic voltammograms of(a)Pt/WO3-CNTs and(b)Pt/CNTs in 0.5 mol·L-1H2SO4solution at a scan rate of 50 mV·s-1

Table 1 Onset potential,EAS,peak current density,and forward anodic to reverse anodic peak current density ratio ofthe different catalysts for methanol oxidation
Besides,it is well known that the ratio of the forward anodic peak current density(If)to the reverse anodic peak current density(Ib),i.e.,If/Ib,suggests a tolerance to carbonaceous species accumulation of catalysts during methanol electro-oxidation.And the high If/Ibindicates excellent oxidation of methanol during the reverse anodic scan and less accumulation of residues on the catalyst.Here the If/Ibratio for Pt/WO3-CNT is about 1.37,which is much higher than that of Pt/CNT catalyst(0.96),showing the Pt/WO3-CNT has a better tolerance to carbonaceous species accumulation.
Fig.8(a)shows the cyclic voltammograms of Pt/WO3-CNTs catalyst with different mass contents of WO3in 0.5 mol·L-1H2SO4+1.0 mol·L-1CH3OH solution.In Fig.8(b),the effects of WO3content on the anodic peak current density and the If/Ibratio are shown.It can be seen that for Pt/WO3-CNTs catalyst,the peak current density of methanol oxidation increases along with the increasing of WO3content because WO3reduces the Pt nanoparticles size and improves the dispersion of Pt nanoparticles on the surface of CNTs.26However,the increasing of WO3amount would lead to a decrease of the electrode conductivity,thus decreases the reaction performance of Pt/WO3-CNTs catalyst on the contrary,finally,the content of WO3is optimized at ca 25%.

Fig.7 Cyclic voltagrammograms of methanol oxidation on(a)Pt/WO3-CNTs,(b)Pt/CNTs,and(c)WO3/CNTs in 0.5 mol·L-1 H2SO4+1 mol·L-1CH3OH solution at a scan rate of 50 mV·s-1
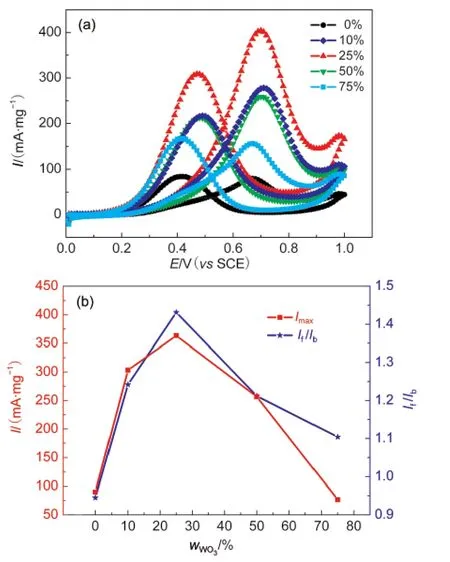
Fig.8 (a)Cyclic voltammograms of the Pt/WO3-CNTs catalyst with different mass contents of WO3in 0.5 mol·L-1H2SO4+1.0 mol·L-1CH3OH solution with a scan rate of 50 mV·s-1;(b)dependencyof anodic peak current density and the ratio ofIf/Ibto the mass fraction of WO3
CO-stripping voltammograms is measured and the characteristic CO stripping curves of Pt/WO3-CNTs and Pt/CNTs catalysts are shown in Fig.9.The onset potential and the peak potential may directly reflect the CO oxidizing ability of the catalysts.It is revealed that the onset potential and the peak potential for the oxidation of adsorbed CO on Pt/WO3-CNTs(Fig.9(a))are much lower than those on Pt/CNTs(Fig.9(b)),so WO3efficiently reduces the overpotential of CO oxidation due to forming HxWO3-OHadsspecies at lower potentials,which is helpful to oxidize COadsthrough bi-functional mechanism.33

Fig.9 CO-stripping voltammograms of Pt/WO3-CNTs and Pt/CNTs catalysts in 0.5 mol·L-1H2SO4solution at room temperature and a scan rate of 50 mV·s-1

Fig.10 Chronoamperomtric curve of Pt/CNTs and Pt/WO3-CNTs in 0.5 mol·L-1H2SO4+1 mol·L-1CH3OH solution at an operation potential of 0.7 V
The chronoamperometry(CA)curves for the three catalysts are shown in Fig.10.These curves reflect the activity and stability of the catalysts to catalyze methanol oxidation.Obviously,the decay in the methanol oxidation current with time varies.But after 100 min the current density of Pt/WO3-CNTs catalyst is 15 times higher than that of Pt/CNTs catalyst.It shows that the modification of WO3can effectively improve the resistance to toxic and stability of Pt-based catalyst for methanol oxidation.This significant improvement in the catalytic performance of the Pt/WO3-CNTs catalysts may be attributed to three factors:first,the Pt and WO3particles supported on the carbon are smaller and more uniformly distributed;second,more metallic Pt is present on Pt/WO3-CNTs than on Pt/CNTs catalyst.Third,in the presence of WO3,the hydrogen adsorbed on the Pt spills over onto the surface of the WO3and forms HxWO3,thus releasing these Pt active sites.Subsequently,HxWO3can be readily oxidized to release hydrogen ions,electrons,and WO3.34,35This cyclic process will accelerate the dehydrogenation of methanol on Pt and improve the catalytic performance of methanol oxidation.The cyclic process on the Pt/WO3-CNTs catalyst is speculated to occur as follows:33,34
4 Conclusions
Nano-WO3modified carbon nanotubes were prepared by the conventional means with sodium tungstate as the precursor.Platinum supported on the WO3-modified CNTs(Pt/WO3-CNTs)was prepared by means of microwave heating ethylene glycol method.Electrochemical analysis shows that the Pt/WO3-CNTs catalysts prepared exhibit excellent catalytic activity and stability for methanol electro-oxidation.
(1) Jung,E.H.;Jung,U.H.;Yang,T.H.;Peak,D.H.;Jung,D.H.;Kim,S.H.International Journal of Hydrogen Energy 2007,32,903.doi:10.1016/j.ijhydene.2006.12.014
(2) Li,X.;Chen,J.L.;Zhu,Z.H.;De Marco,R.;Bradley,J.;Dicks,A.Energy&Fuels 2009,23,3721.doi:10.1021/ef900203h(3)Han,D.M.;Guo,Z.P.;Zeng,R.;Kim,C.J.;Meng,Y.Z.;Liu,H.K.International Journal of Hydrogen Energy 2009,34,2426.doi:10.1016/j.ijhydene.2008.12.073
(4) Corpuz,A.R.;Olson,T.S.;Joghee,P.;Pylypenko,S.;Dameron,A.A.;Dinh,H.N.;OʹNeill,K.J.;Hurst,K.E.;Bender,G.;Gennett,T.;Pivovar,B.S.;Richards,R.M.;OʹHayre,R.P.Journal of Power Sources 2012,217,142.doi:10.1016/j.jpowsour.2012.06.012
(5) Kakati,N.;Lee,S.H.;Maiti,J.;Yoon,Y.S.Surface Science 2012,606,1633.doi:10.1016/j.susc.2012.07.008
(6)Chu,Y.Y.;Wang,Z.B.;Jiang,Z.Z.;Gu,D.M.;Yin,G.P.Journal of Power Sources 2012,203,17.doi:10.1016/j.jpowsour.2011.11.025
(7) Remona,A.M.;Phani,K.L.N.Journal of Fuel Cell Science and Technology 2011,8,011001.
(8) Chu,Y.H.;Shul,Y.G.International Journal of Hydrogen Energy 2010,35,11261.doi:10.1016/j.ijhydene.2010.07.062(9) Wu,G.;Swaidan,R.;Cui,G.F.Journal of Power Sources 2007,172,180.doi:10.1016/j.jpowsour.2007.07.034
(10) Chung,Y.S.;Pak,C.;Park,G.S.;Jeon,W.S.;Kim,J.R.;Lee,Y.;Chang,H.;Seung,D.Journal of Physical Chemistry C 2008,112,313.doi:10.1021/jp0759372
(11) Piela,P.;Eickes,C.;Brosha,E.;Garzon,F.;Zelenay,P.Journal of the Electrochemical Society 2004,151,A2053.
(12) Profeti,L.P.R.;Profeti,D.;Olivi,P.International Journal of Hydrogen Energy 2009,34,2747.doi:10.1016/j.ijhydene.2009.01.011
(13) Zhou,C.M.;Wang,H.J.;Liang,J.H.;Peng,F.;Yu,H.;Yang,J.Chinese Journal of Catalysis 2008,29,1093.doi:10.1016/S1872-2067(09)60007-3
(14) Frolova,L.A.;Dobrovolsky,Y.A.Russian Chemical Bulletin 2011,60,1101.doi:10.1007/s11172-011-0174-z
(15) Guo,D.J.;You,J.M.Journal of Power Sources 2012,198,127.doi:10.1016/j.jpowsour.2011.10.017
(16)Xu,M.W.;Gao,G.Y.;Zhou,W.J.;Zhang,K.F.;Li,H.L.Journal of Power Sources 2008,175,217.doi:10.1016/j.jpowsour.2007.09.069
(17) Shen,P.K.;Chen,K.Y.;Tseung,A.C.C.Journal of the Electrochemical Society 1995,142,L85.
(18) Shen,P.K.;Tseung,A.C.C.Journal of the Electrochemical Society 1994,141,3082.doi:10.1149/1.2059282
(19) Shen,P.K.;Chen,K.Y.;Tseung,A.C.C.Journal of the Chemical Society-Faraday Transactions 1994,90,3089.doi:10.1039/ft9949003089
(20) Cui,X.Z.;Shi,J.L.;Chen,H.R.;Zhang,L.X.;Guo,L.M.;Gao,J.H.;Li,J.B.Journal of Physical Chemistry B 2008,112,12024.
(21) Jayaraman,S.;Jaramillo,T.F.;Baeck,S.H.;McFarland,E.W.Journal of Physical Chemistry B 2005,109,22958.doi:10.1021/jp053053h
(22)Zhang,D.Y.;Ma,Z.F.;Wang,G.X.;Konstantinov,K.;Yuan,X.X.;Liu,H.K.Electrochemical and Solid State Letters 2006,9,A423.
(23) Chen,K.Y.;Shen,P.K.;Tseung,A.C.C.Journal of the Electrochemical Society 1995,142,L185.
(24) Shen,P.K.;Chen,K.Y.;Tseung,A.C.C.Journal of Electroanalytical Chemistry 1995,389,223.doi:10.1016/0022-0728(95)03974-L
(25)Yang,C.Z.;van der Laak,N.K.;Chan,K.Y.;Zhang,X.Electrochimica Acta 2012,75,262.doi:10.1016/j.electacta.2012.04.107
(26)Cui,Z.M.;Feng,L.G.;Liu,C.P.;Xing,W.Journal of Power Sources 2011,196,2621.doi:10.1016/j.jpowsour.2010.08.118
(27) Rajesh,B.;Karthik,V.;Karthikeyan,S.;Thampi,K.R.;Bonard,J.M.;Viswanathan,B.Fuel 2002,81,2177.doi:10.1016/S0016-2361(02)00162-X
(28) Sheng,J.F.;Ma,C.A.;Zhang,C.;Li,G.H.Acta Physico-Chimica Sinica 2007,23,181.[盛江峰,马淳安, 张 诚,李国华.物理化学学报,2007,23,181.]doi:10.3866/PKU.WHXB20070209
(29) Rajeswari,J.;Viswanathan,B.;Varadarajan,T.K.Materials Chemistry and Physics 2007,106,168.doi:10.1016/j.matchemphys.2007.05.032
(30)Ahmadi,R.;Amini,M.K.International Journal of Hydrogen Energy 2011,36,7275.doi:10.1016/j.ijhydene.2011.03.013
(31) Raghuveer,V.;Viswanathan,B.Journal of Power Sources 2005,144,1.doi:10.1016/j.jpowsour.2004.11.033
(32)Su,F.B.;Poh,C.K.;Tian,Z.G.;Xu,G.W.;Koh,G.Y.;Wang,Z.;Liu,Z.L.;Lin,J.Y.Energy&Fuels 2010,24,3727.doi:10.1021/ef901275q
(33) Park,K.W.;Choi,J.H.;Sung,Y.E.Journal of Physical Chemistry B 2003,107,5851.doi:10.1021/jp0340966
(34)Tseung,A.C.C.;Chen,K.Y.Catalysis Today 1997,38,439.doi:10.1016/S0920-5861(97)00053-9
(35) Ye,J.L.;Liu,J.G.;Zou,Z.G.;Gu,J.;Yu,T.Journal of Power Sources 2010,195,2633.doi:10.1016/j.jpowsour.2009.11.055
