烷烃催化制丙烯研究进展
2013-09-15李福超张久顺袁起民
李福超,张久顺,袁起民
(中国石化石油化工科学研究院,北京100083)
乙烯和丙烯是基本有机化工原料,据统计[1],2000—2010年世界市场对乙烯、丙烯需求量的年均增长率为4%~5%,且对丙烯的需求高于乙烯。石脑油蒸汽裂解是生产乙烯和丙烯的传统方法,然而蒸汽裂解装置的反应温度高达800℃,CO2排放量大,产物分布不理想,并且新增的蒸汽裂解装置主要以乙烷为原料,仅能联产少量的丙烯[2],无法满足市场对丙烯的需求,因此该技术的发展逐渐受到制约。烷烃是直馏石脑油的主要组分,我国丰富的液化气中也含有大量的丙烷和丁烷。在当今石油资源供应日趋紧张的形势下,从资源利用与节能环保的角度出发,越来越多的学者致力于烷烃催化制丙烯工艺的研究。催化裂解和催化氧化在降低反应温度的基础上,提高烷烃的反应活性、改善产物分布、达到增产丙烯的目的,成为极具发展潜力的增产低碳烯烃替代工艺。在开发高活性催化剂与认识反应规律方面,国内外科研人员开展了大量的研究工作。本文对烷烃催化裂解与催化氧化的研究情况进行综述,并分析两种路径的技术优势、存在的问题及今后的发展趋势。
1 烷烃催化裂解制丙烯
催化裂解通过引入催化剂,耦合高温热裂化和催化裂化的反应优势,降低裂解反应的活化能,能够将烷烃转化为乙烯、丙烯等低碳烯烃。催化裂解制烯烃的研究起源于20世纪50年代末,针对如何增产低碳烯烃,各国相继开发了一系列催化剂及工艺技术,并对催化裂解的反应规律进行了深入研究。根据目前的研究报道,烷烃催化裂解制丙烯催化剂主要有金属氧化物催化剂和分子筛催化剂。
1.1 金属氧化物催化剂及其反应规律
1.1.1 金属氧化物催化剂 金属氧化物催化剂的水热稳定性较好,多用于管式固定床反应器,最具有研究前景的是钙铝类催化剂和钒酸钾催化剂。金属氧化物催化剂的活性组分主要是碱金属、碱土金属和过渡金属的氧化物或复合盐[3];使用的载体主要有氧化铝、沸石等,载体通常具有一定的反应活性;助剂可以增强催化剂的抗积炭性能,提高催化剂的稳定性。Kikuchi等[4]提出,组成为51.46%CaO和47.73%Al2O3的催化剂反应效果最佳,该催化剂的缺点是CO2产率较高。Lemonidou等[5]评价了一系列金属氧化物催化剂的反应性能,发现12CaO-7Al2O3催化剂的反应效果最优,以正己烷为原料反应时,乙烯产率为34.8%,丙烯产率为19.0%。研究认为,CaO/Al2O3催化剂中的活性氧是催化活性中心,活性氧可以促进催化剂表面甲基自由基的生成,有利于气相中烃类的裂化反应。X衍射结果说明,Ca12Al14O33与Ca3Al3O6晶相的形成有利于提高烯烃产率,这与 Kikuchi等[4]的结论一致。
金属氧化物催化剂的反应温度比蒸汽裂解低30~50℃,乙烯收率远高于丙烯收率[6],主要存在焦炭沉积量大的问题。为了抑制催化剂结焦,比较有效的方法是在催化剂中加入碱金属化合物。Mukhopadhyay等[7]研究发现,将 K2CO3浸渍到12CaO-7Al2O3催化剂中,催化剂结焦量降低,分析原因认为K2CO3不仅能够加快焦炭气化反应和水煤气变换反应的速率,而且能够催化焦炭前躯物如稠环芳烃和焦油的气化。Lemonidou等[5]认为,高温焙烧、低比表面积的CaO/Al2O3催化剂可以有效抑制焦炭的生成。有研究报道[8],KVO3可以降低催化剂表面结焦程度,其最优用量为10%以上。
1.1.2 反应规律 在金属氧化物的作用下,催化裂解能够提高低碳烯烃产率,但这并不是催化反应的效果。研究表明[9],烷烃在金属氧化物催化剂上的裂解反应依然遵循自由基机理。通过催化裂解和热裂解产物选择性的比较可知,在相同的烷烃转化率下,除了焦炭和COx外,其余产物的选择性几乎没有发生变化。根据Rice理论[10],链引发步骤决定原料的转化深度,而产物选择性主要由链增长和链终止步骤决定。因而,催化裂解提高了原料转化率但没有明显改变产物选择性,这可能是由于催化剂影响了链引发步骤。Basu等[11]认为,乙烯产率的提高是由于催化剂提高了链引发步骤自由基的生成速率。Golombok等[12]认为,催化剂颗粒改善了反应系统的热量传递,有利于加速自由基反应。
在认识反应机理的同时,科研人员还对烷烃催化裂解反应动力学进行了大量研究。Lemonidou等[13]以12CaO-7Al2O3为催化剂,研究了正己烷的催化裂解反应。采用Gear算法并结合稳定态假设,建立了包括13种分子和17种自由基,由88个基元反应组成的裂解反应机理模型,并得到催化裂解反应的动力学参数。该模型能够较好地模拟实验结果,当停留时间较短时,以烯烃产物为主;而停留时间较长时,主要生成H2和CO,但是该模型对烷烃和1,3-丁二烯产率的预测值偏低。Pant等[14]考察了正庚烷在 K2CO3浸渍的12CaO-7Al2O3催化剂上的蒸汽裂解性能,并提出了正庚烷裂解的单分子自由基模型,该模型包括1个一级主反应和24个二次反应,催化裂解反应的指前因子和活化能分别为1.7×103m3/(kg·s)和103.8kJ/mol。该模型对甲烷、乙烯和1,3-丁二烯产率的预测值与实验数据非常吻合,丙烯在达到最大值前的模拟效果令人满意。他们还研究了焦炭气化反应,并获得该反应的活化能和反应级数,为抑制催化剂结焦提供了理论依据。
1.2 分子筛催化剂及其反应规律
1.2.1 分子筛催化剂 与金属氧化物催化剂相比,分子筛催化剂在降低反应温度和提高丙烯收率方面更具有优势,目前使用比较广泛的是ZSM-5分子筛。1965年 Mobil公司开发出ZSM-5分子筛[15],并将其应用到催化裂化过程中。ZSM-5分子筛是一种含有十元氧环结构组成两种交叉孔道的MFI型沸石,具有独特的酸性和孔道择形性,能够将烷烃原料选择性裂化为C2~C4烯烃[16],并可以有效抑制氢转移反应。ZSM-5分子筛可以明显提高低碳烯烃收率,当其质量分数达到5%时对低碳烯烃收率的提高程度与反应温度升高150℃相当。采用具有择形性能的小孔分子筛,还可以有效提高丙烯的平衡收率[17]。
研究表明[18-19],ZSM-5分子筛的孔道结构、酸性质和晶粒大小对催化活性有很大影响。由于ZSM-5分子筛在高温水热条件下容易失活,通过对ZSM-5分子筛进行水热处理和金属改性(如磷改性和稀土改性)[20-21],可以有效保护分子筛的结构,调节催化剂的酸性,增强催化剂的水热稳定性,进一步提高丙烯收率。
1.2.2 反应规律 普遍认为,烷烃在酸性催化剂上发生裂解反应时,单、双分子裂化反应机理共同发挥作用。正己烷在分子筛催化剂上的裂解反应路径见图1(HZ表示分子筛)。在链引发阶段,初始正碳离子在单分子裂化反应机理[22]的作用下形成,即催化剂B酸中心上的质子攻击烷烃C—C或C—H键生成非经典的五配位正碳离子,然后经α位断裂生成较小的烷烃分子或氢气和一个与之互补的三配位正碳离子,而生成的三配位正碳离子按照双分子裂化反应机理[23]发生β位断裂和氢负离子转移反应。大量研究结果支持了上述反应机理。Krannila等[24]研究了正丁烷在HZSM-5分子筛上的裂化反应,当转化率接近0时,一次产物的初始生成速率几乎相同,其产物分布与单分子裂化反应机理的预测值一致。龙军等[25]借助分子模拟研究认为,在催化剂酸性中心的作用下,烷烃分子链上的叔碳原子或碳链中心处的C—C键和C—H键容易发生质子化,生成的五配位正碳离子主要以桥式结构存在,在α位C—C键或C—H键处发生共价键异裂的几率最大。
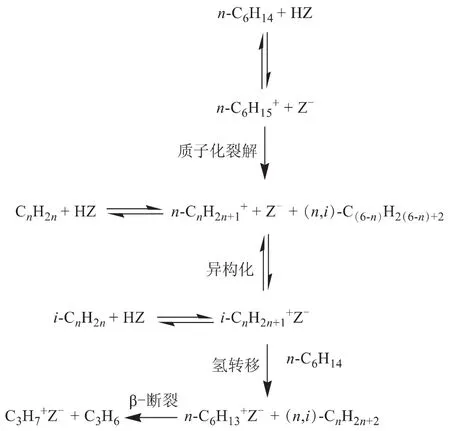
图1 正己烷在分子筛催化剂上的裂解反应路径
在探究MFI分子筛上正己烷的裂化反应机理和动力学方面,Jolly等[26]做了大量的研究工作。在质子化裂解步骤中,分子筛酸性中心的共轭碱发挥重要作用。在高温(大于450℃)条件下,吸附烷烃分子的酸性中心的共轭碱参与质子化裂解反应。在低温条件下,质子化裂解步骤需要第二个活性中心的邻位协助。他们将正己烷分压和分子筛中铝含量的反应级数考虑在内,并结合其他学者的研究结果,分别提出了高温、低温条件下五配位正碳离子的裂解反应机理。Lukyanov等[27]研究了低转化率下(转化率小于30%)正己烷在H-ZSM-5分子筛上的裂化反应,结果表明,分子筛的活性与铝含量成正比,催化活性中心仅是与骨架铝相连的桥羟基。为了确定正己烷质子化裂化和氢转移反应的速率常数,提出了正己烷裂化反应的动力学模型,三种不同硅铝比分子筛上裂化反应的实验结果与模型预测值一致。关于正碳离子物种的结构与性质,文献[26]认为三配位正碳离子以表面烷氧基物种而不是离子物种的形式存在,五配位正碳离子很难以中间物的形式稳定存在于分子筛孔道中,它仅是一种过渡状态的活化络合物。这与Narbeshuber等[28]的观点一致。在表征固体酸催化剂方面,正己烷的裂化反应通常作为分子筛催化剂催化活性的测试反应(α-测试)[27,29]。该方法通过测定在538℃、正己烷初始摩尔分数为13%的条件下,裂化反应的一级动力学速率常数来评价催化剂的活性。在α-测试条件下,氢转移反应对正己烷在HZSM-5分子筛上的裂化影响不明显。
烷烃发生催化裂解反应时,单、双分子裂化反应路径所占的比例受催化剂性质和反应条件(如温度、水油比和烃分压)等因素的影响。由于单分子裂化反应的活化能高于双分子裂化反应,因此高温有利于单分子裂化反应。低转化率、低烯烃浓度也有利于单分子裂化路径[29]。催化剂对两种反应机理影响较大[26],大孔分子筛特别是无定形硅酸铝有利于双分子裂化反应,而中孔的HZSM-5分子筛有利于质子化裂化反应。此外,单分子裂化路径对催化剂B酸中心的酸强度更敏感[30]。在催化裂解过程中,可以用干气作为特征产物来研究单分子裂化反应[31]。胡晓燕等[32]研究认为,正庚烷在新鲜HZSM-5催化剂上发生单分子裂解反应时,产物中氢气、甲烷和乙烷的含量远高于1-庚烯裂解的情况。理论研究表明[33],单分子裂化反应有利于提高液化气中丙烯的体积分数,但干气产率也较高。因此,通过改变反应条件和催化剂结构以合理控制两种裂化路径的反应程度,可以实现增产丙烯同时降低干气产率的目的。
采用分子筛催化剂,催化裂解的反应温度比蒸汽裂解低50~100℃,可以灵活调节丙烯/乙烯产出比,并可大幅减少CO2的排放。此外,采用催化裂解生产低碳烯烃比蒸汽裂解节能21%~30%[34]。目前,催化裂解技术已在工业示范装置上取得突破性进展。SK能源公司和KBR公司共同开发的先进催化制烯烃工艺(ACO)[35],采用Orthoflow反应器系统和ZSM-5高酸性分子筛催化剂,丙烯和乙烯产率之和达65%,丙烯/乙烯质量比为0.8~1.2,实现了高丙烯收率的目标。但是催化裂解的反应温度仍然较高,乙烯收率远低于蒸汽裂解,而且干气和焦炭产率较高,影响了目的产物的总收率。
2 烷烃催化氧化制丙烯
烷烃催化氧化为放热反应,不受热力学平衡的限制,氧气的存在减少了催化剂的积炭,延长了装置的运转周期。因此,烷烃催化氧化制烯烃具有潜在的发展优势,引起了研究人员的高度重视。由于C4以上烷烃氧化脱氢反应产物的选择性难以控制,催化剂表面积炭增多,产物中低附加值CO2所占比例较大,因此对高碳烷烃催化氧化制低碳烯烃的研究较少。近年来研究的热点是丙烷催化氧化脱氢制丙烯。
2.1 催化氧化催化剂
目前,丙烷催化氧化制丙烯催化剂主要包括钒基、钼基、稀土、磷酸盐四类。钒含有部分充满的d轨道,存在+2,+3,+4和+5多种氧化态,不同氧化态的相互转化与氧缺位的形成使钒氧化物广泛应用于选择氧化领域[36]。钒基催化剂的研究主要集中于V-Mg-O和负载型钒基催化剂。
对 V-Mg-O催化剂物相结构的研究表明[37-38],在催化剂表面上镁和钒主要以 Mg2+和V4+形式存在;V含量较低时,丙烯产率较高;当V/Mg比增大时,催化剂的反应活性增强。Schwarz等[39]采用微反应器研究发现,随着钒含量的增加,反应活性逐渐升高,但丙烯选择性降低。Pantazidis等[40]认为,低、中等程度 V 含量的 V-Mg-O催化剂能够提高丙烯收率,MgO影响了催化剂表面的电导率,改变了催化剂的氧化还原性能。
为了提高钒的分散程度,负载型钒基催化剂通常采用比表面积较大的SiO2,TiO2,Al2O3,ZrO2以及磷酸盐等作为载体。催化剂的催化性能取决于载体的性质和钒的负载量。载体的酸碱性可以改变催化剂的表面结构,影响烷烃分子的吸附、中间体的形成、转化以及产物分子的脱附,导致催化剂氧化还原能力的改变。Viparelli等[41]研究了负载在高比表面积TiO2上的V、Nb催化剂的氧化脱氢性能,在低V/Nb比时,V与Nb的相互作用改变了催化剂的表面酸性,提高了催化剂的反应性能。Corma等[42]考察了一系列负载型催化剂的氧化脱氢性能。研究表明,高V含量(质量分数不小于19%)的SiO2,TiO2,Al2O3催化剂的反应活性和选择性几乎与纯V2O5催化剂相同,Sm2O3,MgO,Bi2O3,La2O3负载型催化剂的丙烯选择性较高。分析认为,当V2O5晶相形成时,载体的性质对高V含量负载型催化剂的活性没有很大影响。陈明树等[43]研究认为,载体金属的还原电位越高,催化剂的可还原性越强,丙烷氧化脱氢活性越高;催化剂表面的酸性强弱顺序与丙烷C—H键的活化程度具有较好的相关性,较强的L酸性位易造成深度氧化,较多的碱性位有利于丙烯脱附。
有研究指出[44-45],采用溶胶-凝胶法制备的钒氧化物催化剂,可以使钒在催化剂表面和体相均匀分散,获得比浸渍法更高的催化性能。在催化剂中加入碱金属元素,可以改善负载型催化剂的催化性能。Adamski等[46]研究发现,碱金属元素改变了V2O5/ZrO2催化剂表面钒物种的分散度,有利于单核钒氧根物种的形成。Klisińska等[47]考察了K元素和过渡金属元素(Ni,Cr,Nb,Mo)对VOx/SiO2催化剂反应性能的影响。与过渡金属元素不同,K元素降低了催化剂的反应活性,但是提高产物选择性的能力最突出。他们提出设想,助剂的电负性改变了活性中心的电子密度,影响了催化剂的氧化还原性能。
2.2 催化氧化反应规律
丙烷催化氧化脱氢反应具有平行-顺序反应的特征[48]。当停留时间较短时,丙烷经平行反应生成脱氢和燃烧产物;当停留时间较长时,CO2选择性升高而烯烃选择性降低,CO的选择性也有降低的趋势。气相的丙烷直接与催化剂上的氧物种发生反应,产物在催化剂表面的脱附可以快速完成,丙烷催化氧化脱氢反应按Redox机理进行,其反应步骤如下[49]:
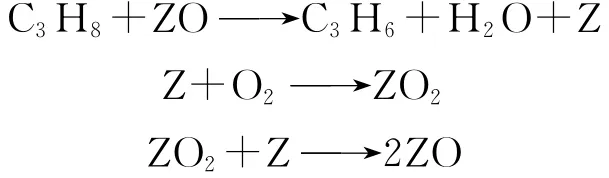
式中:ZO与Z分别代表催化剂的氧化、还原中心。
研究表明,丙烷亚甲基C—H键的活化是催化氧化脱氢反应的速率控制步骤[50]。C—H键有两种活化方式,C—H键均裂生成烷基自由基,C—H键异裂生成烷基负离子。Sokolovskii等[51]分析了烷烃在氧化物催化剂上两种活化途径的可能性,认为C—H键异裂的几率更大,因为均裂活化产生的自由基中心会造成氧自由基物种的生成。烷基负离子不稳定,倾向于释放氢离子生成烯烃,同时转移一个电子到催化剂上,促进丙烷继续脱氢。Burch等[52]也认为,在较低的反应温度(不高于800K)下,对于可还原的过渡金属氧化物催化剂,C—H键活化更倾向于生成烷基负离子。丙烷氧化脱氢反应的活性中心是VO4四面体,但是对于催化剂的活性相仍然存在争论。Kung等[53]认为,正钒酸镁(Mg3V2O8)是生成烯烃的活性组分,烯烃的选择性与分立VO4四面体有关,它主要存在于低钒负载型催化剂中,氧离子以V—O—M形态存在。而Siew等[54]认为,焦钒酸镁(α-Mg2V2O7)是生成烯烃的活性相,具有更高的烯烃选择性。V2O7单元含有V—O—V键,桥氧键合作用较弱,容易从晶格中脱除。焦钒酸镁能够稳定V4+,而V4+的稳定与氧缺位的形成有关。分析产生以上不同结论的原因,可能是由于催化剂原料组成与制备方法存在差异[55]。
催化氧化反应在催化剂氧物种的参与下发生,但是关于活性氧物种的形式以及氧物种的种类仍存在诸多争议。文献[56]报道,V—O—V或V—O—M桥氧参与了丙烷脱氢生成烷基物种的反应,桥氧容易生成C—O键。在不含桥氧物种的Mg3(VO4)2催化剂上,氧化反应的趋势较低。Creaser等[57]采用瞬时实验研究发现,只有晶格氧参与丙烯的形成,可脱附氧引起深度氧化;在相同的丙烷转化率下,在缺少气相氧的条件下,丙烯选择性更高。Grabowski等[58]采用稳态吸附模型(SSAM)研究认为,丙烷和丙烯的反应通过Eley-Rideal机理进行,吸附氧具有游离态的性质,丙烯的生成与烃类的燃烧需要同一种表面氧物种。照日格图等[59]则认为非化学计量氧是丙烷氧化脱氢反应的活性氧物种。催化剂表面的非化学计量氧物种与半导体的类型有关,表面氧物种越多碱性越强。研究人员还尝试采用低活性的氧化物代替高活性的氧分子,以改变催化反应的活性中心,降低COx选择性。Dury等[60-61]研究了向原料中添加少量CO2和N2O对丙烷氧化脱氢性能的影响。结果表明,CO2和N2O的加入,改变了催化剂活性中心的化学状态;CO2并不是非选择性的惰性组分,CO2解离形成吸附氧物种能够使催化剂保持高氧化态,提高了丙烷转化率,但降低了丙烯选择性;N2O抑制了氧气的吸附,提高了催化剂的还原速率,限制了非选择性氧物种的生成,提高了烯烃选择性。
丙烷催化氧化脱氢虽然解决了蒸汽裂解强吸热引起的高温反应,装置结焦周期短等问题,但仍存在不足。丙烯最弱C—H键的键能(360.7kJ/mol)小于丙烷 C—H 键的键能(401.3kJ/mol)[50],丙烷催化氧化时很容易发生深度氧化,生成大量的COx,不仅降低了产物选择性,而且造成产物气体分离的复杂化。因此,丙烷催化氧化脱氢在提高原料转化率与丙烯选择性方面仍需进一步改善。此外,对于催化剂中活性相离子的组成及其电子结构对催化活性的影响以及活性相与周围组分的作用机理尚未认识清楚。
3 结束语
目前,国际市场对丙烯的需求逐年增加,而丙烯产量供不足需。与蒸汽裂解技术相比,烷烃催化制低碳烯烃在降低能耗与资源利用方面具有潜在优势,成为极具发展潜力的增产丙烯替代技术。经过长期研究探索,烷烃催化裂解与催化氧化制低碳烯烃技术已经取得了长足的进展。催化裂解技术原料适应性强,可以灵活调节丙烯/乙烯产出比,产物分布得到较大优化。但是为了获得比蒸汽裂解技术更强的竞争力,需开发新型催化剂体系,以进一步降低反应温度,提高低碳烯烃收率。丙烷催化氧化技术可以充分利用丙烷资源,拓宽丙烯生产路径,并且克服了高温反应的弊端,但是科研人员对催化剂的物相结构和催化作用机理尚存在诸多争论,该技术实现工业化的道路依然很漫长。开发低温高选择性催化剂,加强反应体系的工艺研究,将是丙烷催化氧化的发展趋势。
[1] Park Y K,Lee C W,Kang N Y,et al.Catalytic cracking of lower-valued hydrocarbons for producing light olefins[J].Catalysis Surveys from Asia,2010,14(2):75-84
[2] 曹湘洪.增产丙烯,提高炼化企业盈利能力[J].化工进展,2003,22(9):911-919
[3] 程义贵,茅文星,贺英侃.烃类催化裂解制烯烃技术进展[J].石油化工,2001,30(4):311-314
[4] Kikuchi K,Ishida T,Sakamoto T,et al.A new catalytic cracking process[J].Chem Eng Prog,1985,81(6):54-58
[5] Lemonidou A A,Vasalos I A,Hirschberg E J,et al.Catalyst evaluation and kinetic study for ethylene production[J].Ind Eng Chem Res,1989,28(5):524-530
[6] 刘鸿洲,汪燮卿.ZSM-5分子筛中引入过渡金属对催化热裂解反应的影响[J].石油炼制与化工,2001,32(2):48-51
[7] Mukhopadhyay R,Kunzru D.Catalytic pyrolysis of naphtha on calcium aluminate catalysts.Effect of potassium carbonate impregnation[J].Ind Eng Chem Res,1993,32(9):1914-1920
[8] Jeong S M,Chae J H,Kang J H,et al.Catalytic pyrolysis of naphtha on the KVO3-based catalyst[J].Catal Today,2002,74(3):257-264
[9] Erofeev V I,Adyaeva L V,Ryabov Y V.Pyrolysis of straight-run naphtha on ZSM-5zeolites modified with alkaline-earth metal cations[J].Russian J Appl Chem,2001,74(2):235-237
[10]Kossiakoff A,Rice F O.Thermal decomposition of hydrocarbons,resonance stabilization and isomerization of free radicals[J].J Am Chem Soc,1943,65(4):590-595
[11]Basu B,Kunzru D.Catalytic pyrolysis of naphtha[J].Ind Eng Chem Res,1992,31(1):146-155
[12]Golombok M,Kornegoor M,Van Den Brink P,et al.Surfaceenhanced light olefin yields during steam cracking[J].Ind Eng Chem Res,2000,39(2):285-291
[13]Lemonidou A,Koulouris A,Varvarezos D,et al.Mechanistic model for the catalytic cracking of n-hexane over calcium aluminate catalysts for producing light alkenes[J].Appl Catal,1991,69(1):105-123
[14]Pant K K,Kunzru D.Catalytic pyrolysis of n-heptane:Kinetics and modeling[J].Ind Eng Chem Res,1997,36(6):2059-2065
[15]Degnan T F,Chitnis G K,Schipper P H.History of ZSM-5 fluid catalytic cracking additive development at Mobil[J].Micropor Mesopor Mater,2000,35/36:245-252
[16]Den Hollander M,Wissink M,Makkee M,et al.Gasoline conversion:Reactivity towards cracking with equilibrated FCC and ZSM-5catalysts[J].Appl Catal A:General,2002,223(1/2):85-102
[17]汤效平,周华群,魏飞,等.催化裂解多产丙烯过程热力学分析[J].石油学报(石油加工),2008,24(1):22-27
[18]Corma A,Planelles J,Sánchez-Marín J,et al.The role of different types of acid site in the cracking of alkanes on zeolite catalysts[J].J Catal,1985,93(1):30-37
[19]李丽,高金森,徐春明,等.催化裂解过程及其裂解产物分布的影响因素分析[J].石油与天然气化工,2003,32(6):351-355
[20]Lischke G,Eckelt R,Jerschkewitz H G,et al.Spectroscopic and physicochemical characterization of P-modified H-ZSM-5[J].J Catal,1991,132(1):229-243
[21]朱玉霞,汪燮卿.镧、磷复合添加组分对催化裂化催化剂物化性能的影响[J].石油学报(石油加工),2003,19(4):8-14
[22]Haag W,Dessau R.Duality of mechanism for acid-catalyzed paraffin cracking[C]//Proceeding of the 8th International Congress on Catalysis,Berlin,1984
[23]Greensfelder B S,Voge H H,Good G M.Catalytic and thermal cracking of pure hydrocarbons:Mechanisms of reaction[J].Ind Eng Chem,1949,41(11):2573-2584
[24]Krannila H,Haag W O,Gates B C.Monomolecular and bimolecular mechanisms of paraffin cracking:n-Butane cracking catalyzed by HZSM-5[J].J Catal,1992,135(1):115-124
[25]龙军,魏晓丽.催化裂化生成干气的反应机理研究[J].石油学报(石油加工),2007,23(1):1-7
[26]Jolly S,Saussey J,Bettahar M M,et al.Reaction mechanisms and kinetics in the n-hexane cracking over zeolites[J].Appl Catal A:General,1997,156(1):71-96
[27]Lukyanov D B,Shtral V I,Khadzhiev S N.A kinetic model for the hexane cracking reaction over H-ZSM-5[J].J Catal,1994,146(1):87-92
[28]Narbeshuber T F,Vinek H,Lercher J A.Monomolecular conversion of light alkanes over H-ZSM-5[J].J Catal,1995,157(2):388-395
[29]Haag W,Dessau R,Lago R.Kinetics and mechanism of paraffin cracking with zeolite catalysts[J].Stud Surf Sci Catal,1991,60:255-265
[30]Babitz S,Williams B,Miller J,et al.Monomolecular cracking of n-hexane on Y,MOR,and ZSM-5zeolites[J].Appl Catal A:General,1999,179(1):71-86
[31]Corma A,Orchillés A V.Current views on the mechanism of catalytic cracking[J].Micropor Mesopor Mater,2000,35/36:21-30
[32]胡晓燕,李春义,杨朝合.正庚烷在HZSM-5催化剂上的催化裂解行为[J].物理化学学报,2010,26(12):3291-3298
[33]袁起民,李正,谢朝钢,等.催化裂解多产丙烯过程中的反应化学控制[J].石油炼制与化工,2009,40(9):27-31
[34]刘学龙,张凤秋,周春艳.催化裂解与蒸汽热裂解制烯烃技术经济分析[J].石油化工技术经济,2005,21(4):30-33
[35]Michael J T,Curtis E,Sun C,et al.Naphtha cracking for light olefins production[J].Petroleum Technology Quarterly,2010,15(4):87-91
[36]Haber J.Fifty years of my romance with vanadium oxide catalysts[J].Catal Today,2009,142(3/4):100-113
[37]Gao Xingtao,Ruiz P,Xin Qin,et al.Effect of coexistence of magnesium vanadate phases in the selective oxidation of propane to propene[J].J Catal,1994,148(1):56-67
[38]Corma A,Nieto J M L,Paredes N.Influence of the preparation methods of V-Mg-O catalysts on their catalytic properties for the oxidative dehydrogenation of propane[J].J Catal,1993,144(2):425-438
[39]Schwarz O,Frank B,Hess C,et al.Characterisation and catalytic testing of VOx/Al2O3catalysts for microstructured reactors[J].Catal Communications,2008,9(2):229-233
[40]Pantazidis A,Auroux A,Herrmann J M,et al.Role of acidbase,redox and structural properties of VMgO catalysts in the oxidative dehydrogenation of propane[J].Catal Today,1996,32(1/2/3/4):81-88
[41]Viparelli P,Ciambelli P,Lisi L,et al.Oxidative dehydrogenation of propane over vanadium and niobium oxides supported catalysts[J].Appl Catal A:General,1999,184(2):291-301
[42]Corma A,López-Nieto J M,Paredes N,et al.Oxidative dehydrogenation of propane over supported-vanadium oxide catalysts[J].Stud Surf Sci Catal,1992,72:213-220
[43]陈明树,翁维正,许翩翩,等.酸碱性和氧化还原性对负载型钒基催化剂丙烷氧化脱氢性能的影响[J].天然气化工,1998,23(5):19-22
[44]Sarzi-AmadèM,Morselli S,Moggi P,et al.The effect of solgel promoters on the characteristics of mixed V-Nb oxides and their catalytic properties in propane oxidative dehydrogenation[J].Appl Catal A:General,2005,284(1/2):11-20
[45]Murgia V,Torres E M F,Gottifredi J C,et al.Sol-gel synthesis of V2O5-SiO2catalyst in the oxidative dehydrogenation of n-butane[J].Appl Catal A:General,2006,312:134-143
[46]Adamski A,Sojka Z,Dyrek K,et al.Surface heterogeneity of zirconia-supported V2O5catalysts.The link between structure and catalytic properties in oxidative dehydrogenation of propane[J].Langmuir,1999,15(18):5733-5741
[47]Klisińska A,Samson K,Gressel I,et al.Effect of additives on properties of V2O5/SiO2and V2O5/MgO catalysts:I.Oxidative dehydrogenation of propane and ethane[J].Appl Catal A:General,2006,309(1):10-16
[48]Holzwarth A,Denton P,Zanthoff H,et al.Combinatorial approaches to heterogeneous catalysis:Strategies and perspectives for academic research[J].Catal Today,2001,67(4):309-318
[49]Mamedov E A,Cortés Corberán V.Oxidative dehydrogenation of lower alkanes on vanadium oxide-based catalysts:The present state of the art and outlooks[J].Appl Catal A:General,1995,127(1/2):1-40
[50]Chen K,Iglesia E,Bell A T.Kinetic isotopic effects in oxidative dehydrogenation of propane on vanadium oxide catalysts[J].J Catal,2000,192(1):197-203
[51]Sokolovskii V.Principles of oxidative catalysis on solid oxides[J].Catal Rev:Sci Eng,1990,32(1/2):1-49
[52]Burch R,Swarnakar R.Oxidative dehydrogenation of ethane on vanadium-molybdenum oxide and vanadium-niobium-molybdenum oxide catalysts[J].Appl Catal,1991,70(1):129-148
[53]Kung H H,Kung M C.Oxidative dehydrogenation of alkanes over vanadium-magnesium-oxides[J].Appl Catal A:General,1997,157(1/2):105-116
[54]Siew Hew Sam D,Soenen V,Volta J C.Oxidative dehydrogenation of propane over V-Mg-O catalysts[J].J Catal,1990,123(2):417-435
[55]Holgado M J,San Román S,Malet P,et al.Effect of the preparation method on the physicochemical properties of mixed magnesium-vanadium oxides[J].Mater Chem Phys,2005,89(1):49-55
[56]Michalakos P M,Kung M C,Jahan I,et al.Selectivity patterns in alkane oxidation over Mg3(VO4)2-MgO,Mg2V2O7,and(VO)2P2O7[J].J Catal,1993,140(1):226-242
[57]Creaser D,Andersson B,Hudgins R R,et al.Transient kinetic analysis of the oxidative dehydrogenation of propane[J].J Catal,1999,182(1):264-269
[58]Grabowski R,Słoczyñski J,Grzesik N M.Kinetics of oxidative dehydrogenation of propane over V2O5/TiO2catalyst[J].Appl Catal A:General,2003,242(2):297-309
[59]照日格图,葛庆杰,李文钊,等.Ni-V-O催化剂上丙烷氧化脱氢制丙烯的反应[J].催化学报,2000,21(4):332-336
[60]Dury F,Gaigneaux E M,Ruiz P.The active role of CO2at low temperature in oxidation processes:The case of the oxidative dehydrogenation of propane on NiMoO4catalysts[J].Appl Catal A:General,2003,242(1):187-203
[61]Dury F,Centeno M A,Gaigneaux E M,et al.An attempt to explain the role of CO2and N2O as gas dopes in the feed in the oxidative dehydrogenation of propane[J].Catal Today,2003,81(2):95-105
