基于机器学习的蛋白质相互作用位点预测研究进展
2013-09-05李慧
李 慧
1.金陵科技学院信息技术学院,江苏南京,210001;2.江苏省信息分析工程实验室,江苏南京,210001;3.南京航空航天大学计算机学院,江苏南京,210016
基于机器学习的蛋白质相互作用位点预测研究进展
李 慧1,2,3
1.金陵科技学院信息技术学院,江苏南京,210001;
2.江苏省信息分析工程实验室,江苏南京,210001;
3.南京航空航天大学计算机学院,江苏南京,210016
利用计算方法预测蛋白质之间的相互作用,既省时又省力,弥补了采用原理不同和实验条件限制等因素导致的实验数据具有一定假阳性和假阴性实验的缺陷。针对蛋白质相互作用的功能位点,列出了常用的蛋白质相互作用数据库。为了提高预测的精确度,分析了蛋白质特征属性信息选择,以及常用的机器学习预测方法,总结了蛋白质相互作用位点预测的常用机器学习智能算法模型,给出相应的实验评价标准。
蛋白质相互作用;作用位点;预测;机器学习
1 问题的提出
随着人类基因组计划的完成,生命科学研究已经进入了一个以研究功能基因组为标志的后基因组时代。蛋白质是由基因表达的,因此蛋白质组学的研究成为生物信息学研究的热点之一。蛋白质作为生命活动最主要的体现者和执行者,其间的相互作用不仅在细胞活动中起着关键性作用,而且在各种疾病的治疗方面也具有极大的推进作用。目前,蛋白质之间的相互作用主要从微观层面和宏观层面进行作用研究。在微观上侧重于结合部位[1]的研究,着重研究蛋白质相互作用中的功能位点(界面残基),分析预测蛋白质链上哪些表面残基是界面残基,即发生相互作用的残基。在宏观上侧重于结合对象的研究,着重研究蛋白质之间的相互作用对象及其形成的相互作用网络。
对于蛋白质相互作用的研究通常采用的是生化实验方法,主要包括双杂交技术(two hybrid system)、免疫共沉淀、pull-down技术、交联技术、串联亲和纯化、生物发光共振能量转移技术等方法。这些实验方法虽然能够测定蛋白质之间的相互作用,但由于所采用的原理不同,实验条件限制等因素,导致实验数据具有一定的假阳性和假阴性。随着计算机技术的发展,利用计算方法预测蛋白质之间的相互作用,既省时又省力,弥补实验的缺陷。本文针对蛋白质相互作用的功能位点,列出了常用的蛋白质相互作用数据库;为了提高预测的精确度,分析了蛋白质特征属性信息选择,以及常用的机器学习预测方法,并给出相应的总结思考;最后介绍了常用的实验评估方法,给出下一步的研究方向。
2 蛋白质相互作用数据库
随着生物技术和信息技术的发展,出现了许多与蛋白质相关的数据集。为了更好地对蛋白质进行生物实验技术或计算技术的研究,生物信息学研究者们对这些数据进行分析、组织和整理,按照不同特点形成了不同的数据库。大部分蛋白质相互作用数据库的数据是通过实验方法验证得到的,有些数据库数据集是基于计算方法得到的,有些两者兼有[2]。表1列出了大部分蛋白质相互作用数据库。以下重点介绍其中3个重要的蛋白质相互作用数据库。
2.1 DIP
DIP(Database of Interacting Proteins)数据库由 UCLA的 David Elsenberg实验室于 1999年建立,其蛋白质相互作用数据是通过各种各样的实验获得的。DIP数据库包含内容有蛋白质相关信息、蛋白质相互作用信息和采用的实验技术。截至2013年9月,DIP数据库中收集了 76 270个蛋白质相互作用数据对,其中蛋白质数量为26 071个,涉及的物种数达到了 619个。
2.2 M INT
MIN T(Molecular Interaction Databased)数据库是以结构化形式存取,且是已经发表的通过实验方法验证的蛋白质分子相互作用信息,该数据库不含有遗传或计算方法推断的蛋白质相互作用数据,主要是蛋白质之间的物理相互作用。DIP数据库的最新版本是2011年8月发布的,收集了241 458个蛋白质相互作用数据对,其中蛋白质数量为35 553个,pmids数达到了 5 554个,该数据截至日期是 2013年9月。
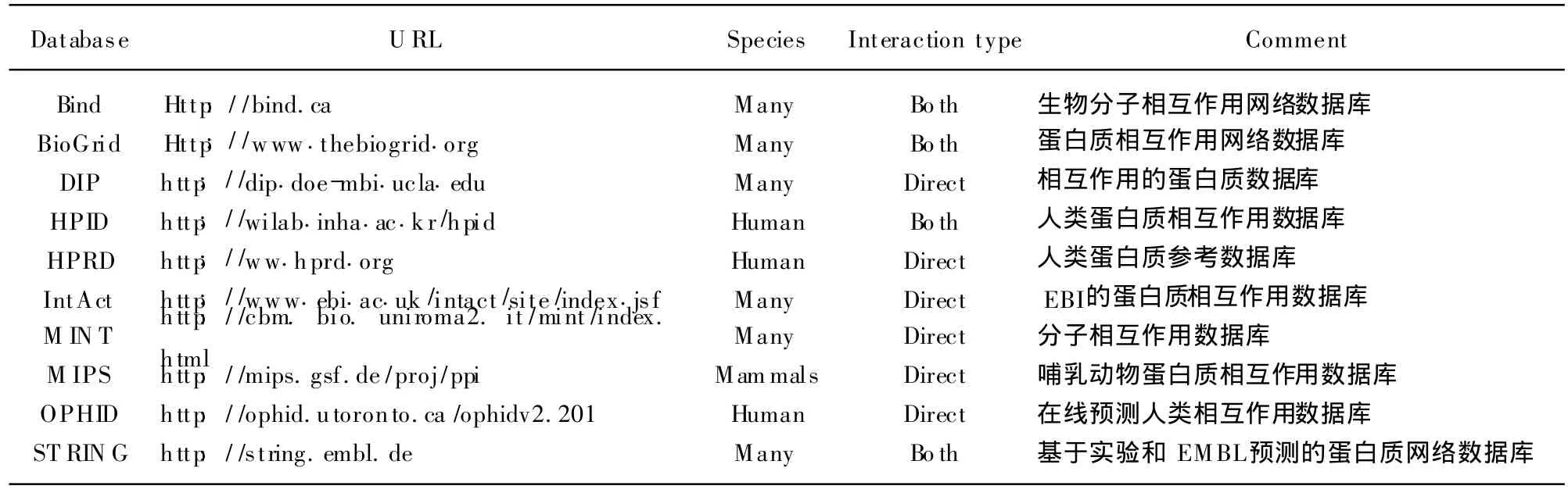
表1 蛋白质相互作用数据库
2.3 BioGRID
BioGRID(Biological General Repository for Interaction Datasets)是一个通过实验方法收集蛋白质遗传和物理的相互作用数据库。该数据库的最新版本是 2013年9月发布的 BioGRID 3.2.104,总计有708 833个蛋白质相互作用数据对,其中蛋白质数量为 52 855个,遗传的相互作用是 269 047个,物理的相互作用是 439 786个。
3 特征属性选择
蛋白质有很多不同的生化特征,为了提高蛋白质相互作用预测的精确度,如何选择合适的特征属性是蛋白质相互作用位点预测的关键。研究者们根据蛋白质相互作用特点,把这些特征大致分成三类:保守性特征、序列特征和结构特征属性[1]。
3.1 保守性特征
生物实验结果已表明,界面残基的保守性比表面残基大。Yan[3]等人采用计算方法对蛋白质相互作用对数据集进行计算,也得出同样结果。保守性表现形式主要有序列谱和残基保守性打分。
序列谱是根据同源序列的多序列比对得到的,可以很好地表达蛋白质系统进化关系。目前,用于蛋白质作用预测的序列谱主要通过 PSI-BLAST和HSSP程序提取出来。 PSI-BLAST进行多序列比对,用矩阵形式表示比对结果得到序列谱。HSSP直接从其hssp文件中提取出蛋白质序列谱。残基保守性打分值有效表达了每个残基的进化保守性。Liang等采用 PSI-BLAST得到序列谱计算保守性打分值[4]。 Landau等人建立ConSurf-HSSP程序计算打分值[5]。
3.2 序列特征
序列特征通过蛋白质的序列信息即可得到,常用的表现形式有疏水性、界面组成及倾向性。通常根据能否与水发生相互作用,将化学物质分为亲水性和疏水性两类。一般原子间仅以非极性共价键相连的分子具有疏水性,它是区分界面残基和非界面残基的一个重要特性。Gallet[6]和Glaser[7]等人研究发现,蛋白质表面中的作用位点残基具有强的疏水性。界面组成及倾向性也是蛋白质相互作用位点预测的一个重要序列特征,Liang等人证实残基的界面倾向性对结果的影响很大[4]。
3.3 结构特征
结构特征通过蛋白质的三维结构信息得到,常用的表现形式有溶剂可及表面积、二级结构。溶剂可及表面积是残基暴露在溶剂中与溶剂相互接触的面积。研究表明,在蛋白质相互作用位点预测中,界面残基比非界面残基具有更大的溶剂可及表面积[8]。对蛋白质的 PDB文件进行 DSSP处理,可以得到该特征值。常用的蛋白质二级结构包括α-螺旋、β-折叠、β-转角和无规则卷曲等结构。研究表明,界面残基中的β-折叠结构比α-螺旋结构更多,其他大致相同[9]。因此,二级结构是蛋白质相互作用位点预测的一个重要特征。
3.4 小结
在进行蛋白质相互作用位点预测时,如何选择合适的蛋白质特征构建特征向量是非常重要的。不同的特征向量使用相同的机器学习算法,产生不同的预测精确度。Lakes Ezkurdia等人使用SVM算法针对不同的特征向量进行预测,得到了不同的预测效果[10]。因此,为了提高蛋白质作用位点预测的精确度,需要根据不同蛋白质数据特点,选择合适的特征属性。另外,还存在一些需要完善的方面。
(1)目前关于蛋白质数据库中的数据信息是有限的,产生足够大和无偏见的数据集需要很长时间。
(2)集成关键结构属性和序列特征属性,形成组合特征。
(3)根据不同蛋白质数据属性特点,对特征向量进行加权处理。
4 作用位点预测智能方法
对蛋白质相互作用位点进行预测的计算方法主要是通过一些机器学习分类器来实现的。首先,对蛋白质源数据进行分析处理,提取出实验数据;其次,选取特征属性并向量化,作为模型的输入参数,构建模型,进行模型训练,不断调整参数直至满足要求;最后,使用该模型预测蛋白质作用功能位点。具体过程如图 1所示。常用的用于蛋白质相互作用位点预测的机器学习方法有贝叶斯、神经网络、支持向量机、隐马尔科夫模型、线性回归、得分函数、条件随机域、随机森林。下面主要介绍3种常用的方法。
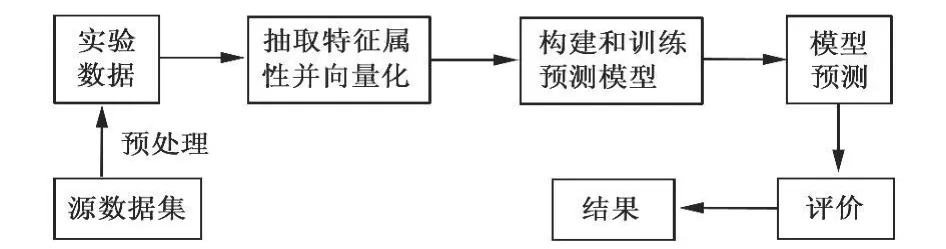
图1 机器学习方法预测蛋白质作用位点过程
4.1 贝叶斯方法
贝叶斯方法是基于不确定性理论进行推理分析的一种方法,能够有效处理不完全或部分数据丢失的数据集。它是一种将先验知识和从数据中收集的新证据相结合的表达模式,已经被广泛应用于蛋白质功能位点预测和相互作用对象预测等生物信息学的各个方面。
蛋白质序列信息样本数据用n维特征向量X={x1,x2,… ,xn}表示,C表示类别,其值Y表示界面残基,值N表示非界面残基。根据贝叶斯定理,分类器公式如下:
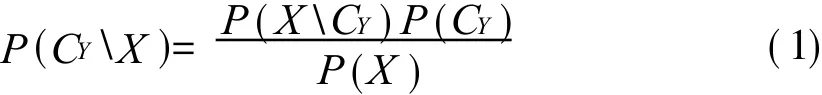
其中,P(CY)表示训练数据集中界面残基的比例,P(XCY)表示属性值为{x1,x2,… ,xn}的界面残基概率值,P(X)表示{x1,x2,… ,xn}在整个数据集中的概率值。如果P(CYX)达到了一个阈值,则认为该残基是界面残基。
朴素贝叶斯分类器要求各属性变量之间相互独立,由式(1)推出式(2)进行分类预测。 Neuvirth[9]和王池社[11]等人使用该方法预测蛋白质界面残基,取得了较好的效果。Bradford等人[12]使用贝叶斯网络分类器进行预测,取得了 82%的成功率。
4.2 神经网络
神经网络是一种模拟生物系统工作的一种方式,具有三层结构:输入层、隐含层和输出层。输入层接受数据的输入,隐含层对输入数据进行非线性变换并处理,最终输出层输出结果。神经网络具有承受噪声数据和对未训练数据分类的能力,已被用于蛋白质相互作用界面残基的预测[13-14]。
蛋白质序列信息样本数据用n维特征向量X={x1,x2,…,xn}表示,被线性映射为一个输入结点,经过中间层非线性变化、处理,输出层输出结果1(界面残基)或 0(非界面残基)。其变化公式为:

式(3)中,IX表示输入,h为隐含层非线性变换函数,f为输出层函数,w1表示输入层和隐含层的连接权重,w2表示隐含层和输出层的连接权重。
文献[15]使用径向神经网络预测蛋白质界面残基,达到了 68.9%的精确度、66.6%的敏感度和67.6%的特异度。文献[16][17]提出了基于PSO优化的径向神经网络预测模型,取得了较好的效果。
4.3 支持向量机
SVM算法使用一种非线性映射,把训练数据集映射到高维空间中,找出最佳超平面把原数据进行归类。SVM能很好地应用于高维数据,避免了维数灾难等问题,现已被广泛应用于生物信息数据的分类问题。
蛋白质序列信息样本数据用n维特征向量X={x1,x2,… ,xn}表示 ,设给定的数据集为 (Xi,Ci),其中Xi是训练元组,具有类标号Ci,Ci∈ {+1,-1},分别对应于 Ci=Yes(界面残基)和Ci=No(非界面残基)。输入数据Xi经过非线性变化,被映射到m维空间Z中。在新空间中,构建非线性决策超平面为 d(Z)=(Z),该非线性超平面对应于原空间中的非线性分类超曲面。文献 [10]给出了不同特征属性组合情况下,使用 SVM分类器分别进行界面残基预测,达到了不同的预测性能。
4.4 小结
到目前为止,大多数机器学习算法已被应用于蛋白质相互作用功能位点预测中,并取得了一定的可行性和可用性。但随着PDB数据库中蛋白质结构信息数据的丰富,改善和提高已有的机器学习算法性能以及开发新的预测算法是新的发展趋势,同时也面临着一个很大的挑战。
(1)不同种类物种蛋白质的信息数据特征不同,应选择合适的机器学习分类算法。如贝叶斯方法能够有效地处理部分数据丢失的数据集。
(2)采用集成机制,组合一些经典算法,充分利用各自算法的优点,进一步提高预测的精确度。
5 实验评估
衡量一个分类器预测性能的好坏,目前还没有一个统一的测量标准,但一般采用下面几种指标来评价其性能。
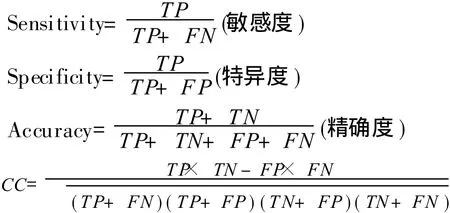
其中,TP表示正确的预测为界面残基的个数;TN表示正确的预测为非界面残基的个数;FP表示错误的预测为界面残基的个数,其实原为非界面残基;FN表示错误的预测为非界面残基个数,其实原为界面残基;CC为相关系数。
6 结束语
蛋白质相互作用的界面残基是生物分子相互作用的基础,也是目前研究的热点,广泛应用于药物靶标识别和新药研制等方面。本文介绍了常用的蛋白质相互作用数据库,综述了蛋白质相互作用特征属性向量的选择以及常用的机器学习预测算法,最后给出常用的评价方法。随着PDB数据库中蛋白质数据的增加,特别是蛋白质结构数据的丰富,如何构造特征向量作为输入参数以及选择合适机器分类器提高预测的精确度是一个值得研究的课题。
[1]Zhou H X,Qin S.Interaction-site prediction for protein complexes:a critical assessment[J].Bioinformatics,2007,23(17):2203-2209
[2]Tuncbag N,Kar G,Keskin O,et al.A survey of available tools and web servers for analysis of protein-protein interactions and interfaces [J]. Briefings in bioinformatics,2009,10(3):217-232
[3]Yan C.Identification of interface residues involved in protein-protein and protein-dna interactions from sequence using machine learning approaches[D].Iowa:Iowa State University,2005:1-120
[4]Liang S,Zhang C,Liu S,et al.Protein binding site prediction using an empirical scoring function[J].Nucleic acids research,2006,34(13):3698-3707
[5]Landau M,Mayrose I,Rosenberg Y,et al.ConSurf2005:the projection of evolutionary conservation scores of residues on protein structures [J].Nucleic acids research,2005,33(Suppl2):299-302
[6]Gallet X,Charloteaux B,Thomas A,et al.A fast method to predict protein interaction sites from sequences[J].Journal of molecular biology,2000,302(4):917-926
[7]Glaser F,Steinberg D M,Vakser I A,et al.Residue frequencies and pairing preferences at protein-protein interfaces [J]. Proteins: Structure, Function,and Bioinformatics,2001,43(2):89-102
[8]Chen H,Zhou H X.Prediction of interface residues in protein-protein complexes by a consensus neural network method:test against NM R data[J].Proteins:Structure,Function,and Bioinformatics,2005,61(1):21-35
[9]Neuvirth H,Raz R,Schreiber G.ProMate:a structure based prediction program to identify the location of protein-protein binding sites[J].Journal of molecular biology,2004,338(1):181-199
[10]Ezkurdia I,Bartoli L,Fariselli P,et al.Progress and challengesin predicting protein-protein interaction sites[J].Briefings in bioinformatics,2009,10(3):233-246
[11]Wang Chishe,Cheng Jiaxing,Su Shoubao,et al.Identification of Interface Residues Involved in Protein-protein Interactions Using Nave Bayes Classifier[J].JournalofFrontiers ofComputer Science and Technology,2009,3(3):293-302
[12]Bradford J R,Needham C J,Bulpitt A J,et al.Insights into protein-protein interfaces using a Bayesian network prediction method[J].Journal of molecular biology,2006,362(2):365-386
[13]Chen H,Zhou H X.Prediction of interface residues in protein-protein complexes by a consensus neural network method:test against NM R data[J].Proteins:Structure,Function,and Bioinformatics,2005,61(1):21-35
[14]Porollo A,Meller J.Prediction-based fingerprints of protein-protein interactions [J]. PROTEINS:Structure,Function,and Bioinformatics,2007,66(3):630-645
[15]Wang B,Chen P,Wang P,et al.Radial basis function neural network ensemble for predicting protein-protein interaction sites in heterocomplexes[J].Protein and Peptide Letters,2010,17(9):1111-1116
[16]Shen X L,Chen Y H.Predicting protein interaction sites based on a new integrated radial basis functional neural network[J].Advanced Materials Research,2011,183:387-391
[17]Chen Y,Xu J,YangB,et al.A novel method for prediction ofprotein interaction sites based on integrated RBF neural networks[J].Computers in Biology and Medicine,2012,42(4):402-407
Q811.4
A
1673-2006(2013)12-0072-04
10.3969/j.issn.1673-2006.2013.12.021
2013-10-22
江苏省高校自然科学研究资助项目(13KJD520005)。
李慧(1981-),女,安徽芜湖人,讲师,博士研究生,主要研究方向:数据挖掘和生物信息。
(责任编辑:汪材印)
