碱金属修饰的MoVTeOx/ SiO2上异丁烷选择氧化制MAL反应研究
2013-09-04孙晓丹张舒冬张信伟尹泽群刘全杰翁维正万惠霖
孙晓丹,金 浩,张舒冬,张信伟,尹泽群,刘全杰,翁维正,万惠霖
(1. 中国石油化工股份有限公司抚顺石油化工研究院, 辽宁 抚顺 113001;2. 固体表面物理化学国家重点实验室, 醇醚酯化工清洁生产国家工程实验室,厦门大学化学化工学院, 福建 厦门 361005)
甲基丙烯醛(MAL)是一种重要的有机化工原料,是生产甲基丙烯酸甲酯(MMA)的重要化学中间体。近年来,由于经济发展的需要和MAL新用途的不断发现,以及合理利用资源的要求,异丁烷选择氧化制 MAL已成为研究热点[1-3]。虽然目前这种方法由于MAL产率较低而不能用于工业生产,但从长远来看,该方法由于具有工艺相对简单,对环境比较友好等优点,越来越受到人们的重视。
异丁烷选择氧化制甲基丙烯醛(MAL)需要转移六个电子,反应中需要催化剂有适当的氧化还原性和酸碱性,不仅要有负责使异丁烷氧化脱氢的活性中心,而且还需要有负责插氧的活性中心。因此,目前报道的异丁烷选择氧化制 MAL催化剂主要是杂多化合物体系和MoVTeOx复合氧化物体系[4-8],都含有 V和(或)Mo元素,但负载型 MoVTeOx催化剂上异丁烷选择氧化制MAL反应的研究,迄今很少报道。我们在前期的研究工作中,采用常规浸渍法制备的 SiO2和 SBA-3等负载的MoV0.8Te0.23Ox催化剂在异丁烷选择氧化制MAL的反应中表现出较好的催化性能[9]。据文献报道,对于丙烷选择氧化制丙烯酸反应,在MoVSbOx催化剂体系上,碱金属K的加入可以改善丙烯酸的选择性,一般认为碱金属通过改变催化剂表面性质和酸性位数目而提高丙烯酸的选择性[10,11]。Ivars等[12]报道了在MoVSbOx催化剂上丙烷氧化制丙烯酸的反应中,碱金属的加入提高了丙烯酸的选择性却降低了乙酸的选择性。加入的碱金属电负性越低,丙烯酸的选择性越高,但当加入碱金属Cs的时候,催化剂的表现却不遵循该规律,得到的丙烯酸选择性比预期的要低,这可能与催化剂中形成的Cs-M1相有关。对于同一催化剂体系,碱金属效应还取决于烷烃的种类。Nieto等[13]报道了在VOx/Al2O3催化剂上乙烷和丙烷的氧化脱氢反应,研究发现K的加入提高了丙烯的选择性却降低了乙烯的选择性。由此可见,碱金属影响烷烃氧化产物选择性的原因尚不清楚,有待深入研究。
本文以SiO2负载的MoV0.8Te0.23Ox催化剂为基础,采用色谱-微反技术考察了碱金属(Li、Na、K和Cs)的添加对催化剂性能的影响,以期获得反应性能较好的碱金属修饰的异丁烷选择氧化制 MAL催化剂。
1 实验部分
1.1 催化剂的制备
采用共浸渍法制备催化剂。催化剂的制备过程:按 Mo/V/Te/M=1︰0.8︰0.23︰m(M 为碱金属Li、Na、K和Cs,m为碱金属与Mo的摩尔比)的摩尔比例,称取一定量钼酸铵、偏钒酸铵、碲酸及相应的碱金属碳酸盐(其中碱金属锂所用的前躯体盐为硝酸锂),配成水溶液。称取一定量的载体SiO2,在上述溶液中浸渍12 h后在油浴(80 ℃)上蒸干水分,然后放入马弗炉中在空气气氛下600 ℃焙烧4 h,即制得负载型 MoV0.8Te0.23MmOx/SiO2催化剂,各催化剂中(Mo+V+Te)/Si的摩尔比均为3%。
1.2 催化剂的反应评价
催化剂的反应性能评价在常压固定床微型反应装置上进行,反应器由直径为8 mm的石英管制成,催化剂用量200 mg(40~60目)。为避免发生气相反应,反应器的剩余空间装满石英砂颗粒(25~50目)。原料气(i-C4H10︰O2︰N2=1︰1︰4)和产物用两台气相色谱仪在线分析(GC-2010,GC-14C,日本岛津),反应尾气在进入色谱取样阀之前加热至150 ℃,以免产物冷凝。i-C4H10、i-C4H8、C3H6、CO以及 CO2等组份经涂渍角鲨烷的Al2O3柱(柱温70 ℃)和碳分子筛柱(柱温70 ℃)分离后由TCD检测;甲基丙烯醛(MAL),丙酮,丙醛等含氧有机物经RT-PLOTQ柱(柱温150 ℃)分离后由FID检测。
1.3 催化剂的常规表征
催化剂的比表面积、孔容及孔径分布测试在Micromeritics Tristar 3020型全自动物理化学吸附仪上完成。样品首先在120 ℃抽真空预处理1 h,再升温至300 ℃处理3 h。以氮气为吸附质,在液氮温度下进行吸附。比表面积测试采用的是多层物理吸附方法(即BET方程),而孔容及孔径分布数据采用BJH方法计算(脱附曲线)。
X-射线粉末衍射实验在 Panalytical公司的X’Pert Pro型X-射线粉末衍射仪上进行。仪器的工作条件为:管电流30 mA;管电压40 kV;X’Celerator超能阵列探测器,以Cu-Kα(λ=0.154 06 nm)为辐射源。测试范围(2θ)为 10~90 deg.,采用X’celerator_normal方式进行测试,扫描步长 0.016 deg./step,每步时间 20 s。所得谱图均经过 X’pert Highscore软件处理后,进行解谱分析。
拉曼光谱测试在 Renishaw inVia Raman Microscope显微拉曼光谱仪上进行,仪器分辨率为2 cm-1,以532 nm线作为激发光源,激光输出功率为23 mW,采用Leica PL Fluotar 50×物镜,CCD检测器。扫描范围为200~2 000 cm-1。
程序升温还原实验装置由气体净化系统,GC-950气相色谱仪,海欣色谱工作站 V4.0,连续流动微型反应器,小型管式电阻炉和AI-708P型程序控温仪组成。还原过程中生成的H2O,通过置于液氮调制的乙醇固-液混合物中的冷阱除去。催化剂用量50 mg(40~60目)。每次TPR实验前催化剂先在 500 ℃下用 O2/Ar(20 mL/min,5% O2)混合气处理 30 min,待样品温度降至室温后将样品切入H2/Ar(20 mL/min,5% H2)混合气,按一定升温程序在H2/Ar气氛下升温到50 ℃预处理60 min,待基线平稳后,按 10 ℃/min的升温速率从 50 ℃升至800 ℃,并用TCD在线检测尾气,色谱信号由海欣色谱工作站收集并在线处理。
2 不同碱金属的添加对 3%MoV0.8Te0.23Ox/SiO2催化剂反应性能的影响
由表1可以看出,3% MoV0.8Te0.23Ox/SiO2催化剂在经相同含量的碱金属修饰后,异丁烷的转化率随碱金属原子序数的增加先减小后增大;MAL的选择性则呈先升高后降低的趋势变化,其中,在MoV0.8Te0.23Na0.2Ox/SiO2催化剂上MAL的选择性最高,为 61.8%;COx的选择性呈先减小后增大的趋势,而异丁烯的选择性则在8%~12%之间波动。从表中的数据可以看出,在所加入的4种碱金属中,铯的添加使3% MoV0.8Te0.23Ox/SiO2催化剂上MAL的收率由2.0%提高到了2.9%,起到了促进作用。
3 催化剂的表征结果与讨论
3.1 MoV0.8Te0.23M0.2Ox/SiO2催化剂的BET表征
从表2可以看出,当3% MoV0.8Te0.23Ox/SiO2催化剂中加入相同含量的碱金属时,催化剂的比表面积均显著降低,平均孔径增大。催化剂比表面积减小的原因可能是原来多孔性SiO2的孔洞被Mo、V、Te和碱金属的氧化物晶粒占据后,真实表面减少而且所得催化剂密度增加。由于比表面积是以催化剂质量为基准(m2·g-1),单纯密度的提高也将导致测量所得的比表面积减小。MoV0.8Te0.23M0.2Ox/SiO2系列催化剂中,MoV0.8Te0.23Cs0.2Ox/SiO2催化剂的比表面积最大,平均孔径最小,这可能与相同的反应条件下,该催化剂的性能最好有一定的关系。
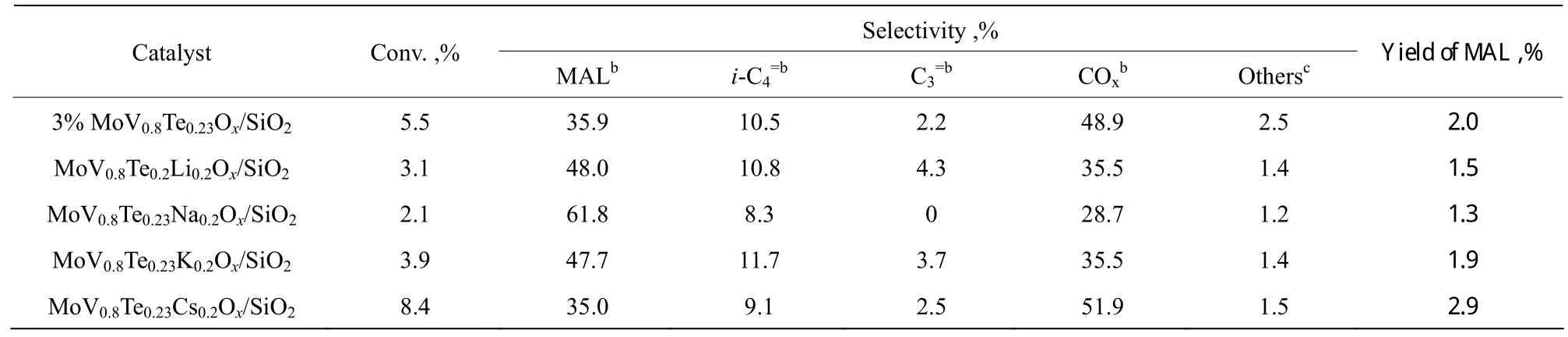
表1 相同含量碱金属的添加对3% MoV0.8Te0.23Ox /SiO2催化剂反应性能的影响aTable 1 Catalytic performance of MoV0.8Te0.23M0.2Ox /SiO2a

表2 MoV0.8Te0.23M0.2Ox/SiO2催化剂的比表面积和平均孔径Table 2 Physical characteristics of MoV0.8Te0.23M0.2Ox/SiO2
3.2 MoV0.8Te0.23M0.2Ox/SiO2催化剂的XRD表征
图 1是经相同含量碱金属修饰的MoV0.8Te0.23M0.2Ox/SiO2催化剂的XRD图。
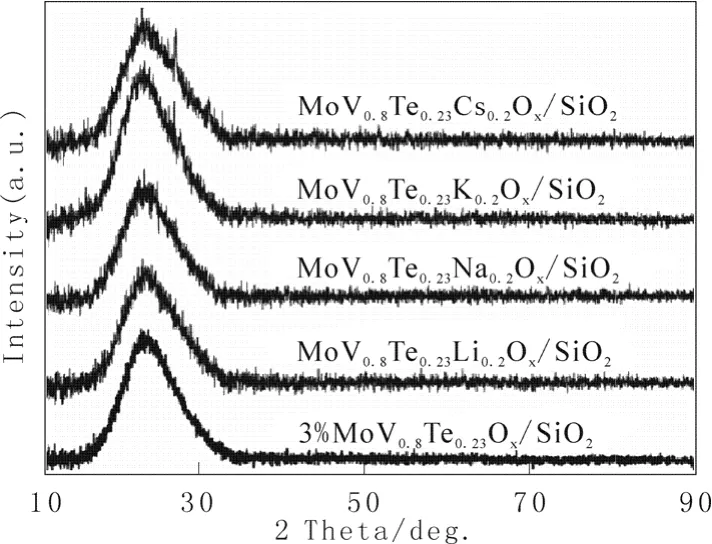
图1 MoV0.8Te0.23M0.2Ox/SiO2催化剂的常规XRD图Fig.1 XRD patterns for MoV0.8Te0.23M0.2Ox/SiO2 catalysts
由图1可知,除了MoV0.8Te0.23Cs0.2Ox/SiO2催化剂之外,其他催化剂只在 2θ=14°~33°范围出现了载体SiO2的特征衍射峰,未检测到与钼氧、碲氧或钒氧物种相关的衍射峰,表明这些催化剂上钼氧、碲氧、钒氧物种或其复合氧化物可能都以高分散或无定形状态存在。MoV0.8Te0.23Cs0.2Ox/SiO2催化剂在 2 θ=25.9°和 29.8°处出现了归属于晶相 TeO2的衍射峰(JCPDS, 65-2825)。这可能是因为原来催化剂中一些无定形碲氧物种随着 Cs的加入转变成晶相TeO2所致。在反应过程中,TeO2中的Te-O活性位被还原后能快速得到体相晶格氧的补充,从而促进晶格氧的传递,这也是添加Cs的催化剂反应性能有所提高的原因之一。
3.3 MoV0.8Te0.23M0.2Ox/SiO2催化剂的 Raman光谱表征
图2是MoV0.8Te0.23M0.2Ox/SiO2催化剂的拉曼谱图。MoV0.8Te0.23M0.2Ox/SiO2催化剂都在 1034 cm-1附近出现了拉曼峰,且该谱峰位置并未因添加碱金属的种类不同而发生明显改变,该拉曼峰可归属为直接与载体表面键合的分立O=V(-O-Si)3的V=O键伸缩振动,该物种也是烷烃氧化脱氢的活性物种之一[14,15]。由图可以看出,随着碱金属的加入,3%MoV0.8Te0.23Ox/SiO2催化剂上位于948 cm-1附近、归属为六配位Te多钼酸盐物种的特征峰强度有所减弱[13,16],855 cm-1附近归属于β-MoO3的肩峰逐渐偏移至817 cm-1附近[17-19]。MoV0.8Te0.23M0.2Ox/SiO2(M=Li, Na,K)催化剂都在 817 cm-1附近出现了归属于晶相α-MoO3的拉曼峰,尤其是MoV0.8Te0.23Li0.2Ox/SiO2催化剂,其在991 cm-1附近也出现了归属于晶相α-MoO3的拉曼峰[17,20]。由 Raman表征可知,3%MoV0.8Te0.23Ox/SiO2催化剂经相同含量碱金属 Li、Na和K的修饰后,不利于Te多钼酸盐在SiO2表面的分散,导致Te多钼酸盐物种结构被破坏,而此时被释放的钼氧物种则倾向于以稳定结构的 MoO3形式存在。MoV0.8Te0.23Cs0.2Ox/SiO2催化剂的拉曼谱图与修饰前的3% MoV0.8Te0.23Ox/SiO2催化剂相比,虽未发生很明显的变化,但在978 cm-1附近出现了归属于单核MoOx物种中Mo=O键的伸缩振动峰[17,21],由此可见,Cs的加入促进了Mo物种在催化剂表面的分散。
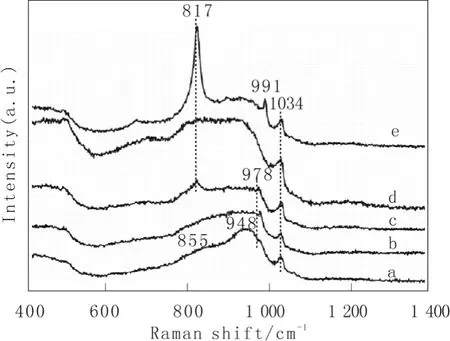
图2 MoV0.8Te0.23M0.2Ox/SiO2催化剂的Raman谱Fig.2 Raman spectra of the catalysts
3.4 MoV0.8Te0.23M0.2Ox/SiO2催化剂的H2-TPR表征
图 3是加入相同含量碱金属的MoV0.8Te0.23M0.2Ox/SiO2催化剂的H2-TPR图。

图3 MoV0.8Te0.23M0.2Ox/SiO2催化剂的H2-TPR图Fig.3 H2-TPR profiles of the catalysts
3% MoV0.8Te0.23Ox/SiO2催化剂在 592 ℃和 653℃处出现两个还原峰,前者与VOx/SiO2催化剂的还原峰温相对应,结合文献[22,23]可将其归属为 V5+→V3+的还原过程,后者可归属为Mo6+→ Mo4+的还原过程。结合前文的拉曼表征结果可知,前者对应于VO4物种的还原,后者可归属为多钼酸盐物种的还原。经相同含量碱金属修饰后,催化剂上592 ℃和653 ℃处的还原峰位向低温方向偏移,可以观察到MoV0.8Te0.23M0.2Ox/SiO2催化剂在616 ℃、529 ℃和489℃附近出现了新的还原峰,同时592 ℃左右的还原峰强度下降。其中,489 ℃附近出现的归属于碲氧物种的还原峰[24],表明催化剂在添加碱金属后,出现易还原的碲氧物种,提高了催化剂的可还原性能。
4 结束语
基于上述实验结果我们可以得出以下结论:在3% MoV0.8Te0.23Ox/SiO2催化剂中添加相同含量的碱金属后,只有Cs的添加对催化剂的反应性能起到了促进作用。3% MoV0.8Te0.23Ox/SiO2催化剂中一些无定形碲氧物种随着Cs的加入转变成晶相TeO2,在反应过程中,TeO2中的Te-O活性位被还原后能快速得到体相晶格氧的补充,从而促进晶格氧的传递,这也是添加 Cs的催化剂反应性能有所提高的原因之一。在接下来的工作中,我们将进一步考察Cs含量对3% MoV0.8Te0.23Ox/SiO2催化剂性能的影响。
[1] Guan J Q, Song K, Xu H Y, Wang Z Q, et al. Oxidation of isobutane and isobutene to methacrolein over hydrothermally synthesized Mo-V-Te-O mixed oxide catalysts[J]. Catalysis Communication, 2009,10(5): 528-532.
[2] 朱万春,孙方龙,常加贵,李学福,王国甲. 磷钼杂多化合物催化剂上异丁烷的选择氧化[J]. 石油化工,2003,32(8):641-645.
[3] Costine A, Hodnett B K. Factors limiting selectivity in C3 and C4 amm(oxidation) reactions[J]. Applied Catalysis A: General, 2005,290(1-2): 9-16.
[4] Min J S, Mizuno N. Iron as an effective additive for enhancement of catalytic performance of cesium hydrogen salt of molybdophosphoric acid for selective oxidation of isobutane, propane, and ethane under oxygen-rich and -poor conditions and the catalyst design[J]. Catalysis Today, 2001, 66(1): 47-52.
[5] Sultan M, Paul S, Fournier M, Vanhove D. Evaluation and design of heteropolycompand catalysts for the selective oxidation of isobutane into methacrylic acid[J]. Applied Catalysis A: General, 2004, 259(2):141-152.
[6] Huynh Q, Schuurman Y, Delichere P, et al. Study of Te and V as counter-cations in Keggin type phosphomolybdic polyoxometalate catalysts for isobutane oxidation[J]. Journal of Catalysis, 2009, 261(2):166-176.
[7] Guan J Q, Jing S B, Wu S J, et al. Selective oxidation of isobutane over Mo-V-Te mixed oxide catalysts with different tellurium contents[J].Reaction Kinetics and Catalysis Letters, 2007, 90(1): 27-33.
[8] Guan J Q, Wu H S, Song K, et al. Selective oxidation of isobutane over hydrothermally synthesized Mo-V-Te-Sb-O mixed oxide catalysts[J].Catalysis Communication, 2009, 10(10): 1437-1440.
[9] Sun X D, Yi X D, Hua W Q, et al. Selective oxidation of isobutane to methacrolein over MoVTe mixed oxide supported on SBA-3 and SiO2[J]. Fuel Processing Technology, 2011, 92(8): 1662-1669.
[10] Ivars F, Solsona B, Botella P, et al. Selective oxidation of propane over alkali-doped Mo-V-Sb-O catalysts[J]. Catalysis Today, 2009,141(3-4): 294-299.
[11] Blasco T, Nieto J M J. Oxidation dehydrogenation of short chain alkanes on supported vanadium oxide catalysts[J]. Applied Catalysis A:General, 1997, 157(1-2):117-142.
[12]Corma A, Lopez-Nieto J M, Paredes N, et al. Oxidation dehydrogenation of propane over supported-vanadium oxide catalysts[J]. Studies in Surface Science and Catalysis, 1992, 72: 213-220.
[13] Solsona B, Vazquez M I, Ivars F, et al. Selective oxidation of propane and ethane on diluted Mo-V-Nb-Te mixed-oxide catalysts[J]. Journal of catalysis, 2007, 252(2): 271-280.
[14] Blasco T, Nieto J M L. Oxidation dehydrogenation of short chain alkanes on supported vanadium oxide catalysts[J]. Applied Catalysis A:General, 1997, 157(1-2): 117-142.
[ 15] Corma A, López-Nieto J M, Paredes N, et al. Oxidative dehydrogenation of propane over supported-vanadium oxide catalysts[J]. Studies in Surface Science and Catalysis, 1992, 72:213-220.
[16] Mestl G, Srinivasan T K K. Raman spectroscopy of monolayer-type catalysts: supported molybdenum oxides[J]. Catalysis Review -Science Engineering, 1998, 40: 451-570.
[17] Williams C C, Ekerdt J G, Jehng J M, et al. A Raman and ultraviolet diffuse reflectance spectroscopic investigation of silica-supported molybdenum oxide[J]. Journal of Physical Chemistry, 1991, 95(22):8781-8791.
[18] Holmes S A, Al-Saeedi J, Guliants V V, et al. Solid state chemistry of bulk mixed metal oxide catalysts for the selective oxidation of propane to acrylic acid[J]. Catalysis Today, 2001, 67(4): 403-409.
[19] Wachs I E, Jehng J M, Ueda W. Determination of the chemical nature of active surface sites present on bulk mixed metal oxide catalysts[J].Journal of Physical Chemistry B, 2005, 109(6): 2275-2284.
[20]Al-Saeedi J N, Guliants V V. High-throughput experimentation in multicomponent bulk mixed metal oxides: Mo-V-Sb-Nb-O system for selective oxidation of propane to acrylic acid[J]. Applied Catalysis A:General, 2002, 237(1-2): 111-120.
[21] Banares M A, Wachs I E. Molecular structures of supported metal oxide catalysts under different environments[J]. Journal of Raman Spectroscopy, 2002, 33(5): 359-380.
[22] Cordero R L, Agudo A L. Temperature-programmed reduction and zeta potential studies of the structure of MoO3/Al2O3and MoO3/SiO2catalysts effect of the impregnation pH and molybdenum loading[J].Applied Catalysis, 1991, 74(1): 125-136.
[23] Arena F, Frusteri F, Parmaliana A. How oxide carriers affect the reactivity of V2O5catalysts in the oxidative dehydrogenation of propane[J]. Catalysis Letters, 1999, 60(1-2): 59-63.
[24] 何益明. 丙烷选择氧化制丙烯醛 Mo(V)BiTeO/SiO2催化剂的研究[D]. 厦门:厦门大学,2006.
