CdS 形貌可控制备及其可见光分解水产氢性能
2013-08-20李曹龙赵宇婷上官文峰
李曹龙 赵宇婷 曹 菲 王 飞 王 越 袁 坚 上官文峰
(1 中国药科大学理学院无机化学教研室,南京 211169)
(2 上海交通大学机动学院燃烧与环境技术中心,上海 200240)
0 引 言
能源危机和环境恶化是制约未来人类社会发展的重大问题。目前,世界大多数国家都在开发清洁的可再生能源以应对此挑战。自1972 年Honda-Fujishima 效应的发现[1],揭开了光催化分解水制氢气的序幕,使利用可再生的太阳能从水中获得氢能成为一条具有发展潜力的新途径。
然而至今,光解水制氢技术主要存在光量子效率低的问题,科技工作者试图从各种途径提高其光催化效率[2-4]。如:寻找可见光响应的催化剂,或者着眼于探讨光解水过程的影响因素和机理,以期获得提高光解水效率的途径。文献报道光催化剂的微观形貌和晶体结构对光解水产氢过程有显著影响。CdS 由于具有很强的可见光响应作为标杆材料被大量研究[5-6]。Domen 等[7]以介孔氧化硅作为硬模板,制备结晶度高的CdS 纳米线阵列,可见光下产氢活性很高。通过自组装制备比表面积较大的纳米片状CdS 以及管径在3 nm 左右的空心CdS 纳米棒[8],在波长大于420 nm 可见光照射下,分解水产氢性能得到很大的提高。然而,仅仅有限篇幅文献报道对于解释光解水过程中微观结构与宏观活性的关系还远远不够,有必要获得更多的实验数据予以支持。纳米粒子的表面效应引起CdS 纳米微粒表面电子输运和构型的变化,同时也引起表面电子自旋构象和电子能谱的变化,对其光学、 电学性质等具有重要影响。
另外,有文献报道非晶态的CdS 具有更高的活性,也有报道结晶完整的CdS 具有更好的稳定性。为获得高活性的CdS 催化剂,构筑微观形貌与活性的关系,各种各样微观形貌的CdS 材料,如:空心球状[9-10],多孔膜[11]、纳米球蛇形链状[12]、微笼状[13]以及壳核结构[14]等CdS 都有报道。遗憾的是,绝大部分报道主要是针对某单一的形貌结构与活性关系进行研究,由于光解水制氢的反应条件和设备存在差异,故得到的结果往往很难构建统一的形貌与活性之间的构效关系。
本文以GSH 作为硫源和结构导向剂,通过调节反应物质的nCd/nS比、控制水热温度等制备了CdS 实心纳米球、空心纳米球以及纳米棒。在相同的实验条件和反应参数下对比研究它们在可见光下分解水产氢性能,并对其进行表征,探究并构筑微观形貌与宏观活性之间的构效关系。
1 实验部分
1.1 不同形貌CdS 的制备
空心球状纳米CdS 制备:将0.304 g GSH(国药集团化学试剂有限公司, 分析纯)加入到45 mL 去离子水中搅拌至完全溶解;将0.305 g Cd(NO3)2·4H2O(国药集团化学试剂有限公司,分析纯)按照nCd/nS比1:1 加入上述的溶液中,搅拌30 min;再将上述混合溶液移入80 mL 聚四氟乙烯的高压反应釜中,在160 ℃水热24 h;冷却至室温,先用乙醇反复洗涤,再用去离子水洗涤,目的是除去CdS 表面包裹的有机物以及其它离子;最后在80 ℃下烘干。并将其标记为h-CdS。调节水热温度至120 ℃制备出CdS 实心球状纳米材料,将其标记为s-CdS;以及水热温度至200 ℃制备出CdS 纳米棒状材料,标记为r-CdS。
1.2 催化剂表征
XRD 分析在日本理学RigakuX 射线粉末衍射仪(Cu Kα 靶,λ=0.154 06 nm, 40 kV,20 mA)进行X射线粉末衍射表征。透射电镜分析在日本电子株式会 社/英 国 OXFORD 公 司 JEM-2010/INCA 型HRTEM 上进行。场发射扫描电镜在美国FEI 公司/英 国OXFORD 公 司FEI SIRION 200 型FESEM 上进行。紫外-可见漫反射(DRS)分析在日本岛津公司UV-2450 紫外-可见分光光度计上进行。荧光/磷光/发光分光光度计在美国Perkin Elmer, Inc. 公司LS 50B 型PL 上进行。采用美国Quanta Chrome 公司NOVA1000 进行BET 测试。
1.3 光催化剂的活性评价
光解水产氢性能测试在自制的真空上照式石英玻璃反应器(350 mL)中进行。反应前将光催化剂样品、水和牺牲剂加入到反应器中,然后将反应器抽成真空。反应中利用磁力搅拌使得光催化剂更好的分散,并用冷却水维持反应体系处于室温(20~25 ℃)。光源采用北京畅拓科技有限公司500 W 氙灯,420 nm 以下的光被滤波片滤去,生成的气体由气相色谱定量检出(华爱色谱9160,TCD 检测器,5A 分子筛填充柱,氩气为载气)。反应条件:催化剂的量为0.15 g、40 mL H2O、10 mL 0.35 mol·L-1Na2SO3、10 mL 0.25 mol·L-1Na2S 和5 mL Pt 含量为0.1 mg·mL-1的H2PtCl6·6H2O。其中,加入H2PtCl6·6H2O 的目的是通过原位光沉积法在主催化剂上负载Pt 起到产氢活性位的作用。Na2SO3和Na2S 作为牺牲剂,消耗掉空穴,防止CdS 发生光腐蚀。
2 结果与讨论
2.1 s-CdS、h-CdS 和r-CdS 光催化剂的结构
从图1 给出的X-射线衍射图可知,h-CdS、s-CdS和r-CdS 都是六方相纤维锌矿晶型结构,空间群P63mc,晶格点阵常数是a=0.414 2 nm,c=0.672 4 nm (PDF No. 02-0549)。s-CdS 样 品 对 应 的(002)、(110)、(103)和(112)晶面比较宽,甚至相邻的晶面互相覆盖,可能由于s-CdS 样品在微结构上粒径很小或者结晶度差导致。随着制备温度的提高,CdS 形貌也发生改变,变成空心球和棒状,X-射线衍射图发现晶面变得比较窄,且强度变强,但晶型结构没有变化。(002)晶面随温度增加相对而言高得多的强度和窄的半峰宽说明CdS 晶粒是沿c 轴取向优先定向生长,且晶体生长的取向由无序变成有序,从而实现形貌可控。Gong 等[13]报道也阐述了CdS 晶粒沿c 轴取向排列生长成微观的棒状结构。
2.2 s-CdS、h-CdS 和r-CdS 微观形貌的结构
图2 是s-CdS 样品透射和扫描电镜照片。从图2a 和图2e 可以看出120 ℃条件下制备的CdS 样品为分散性良好的实心纳米球,球的直径主要集中在100~150 nm。
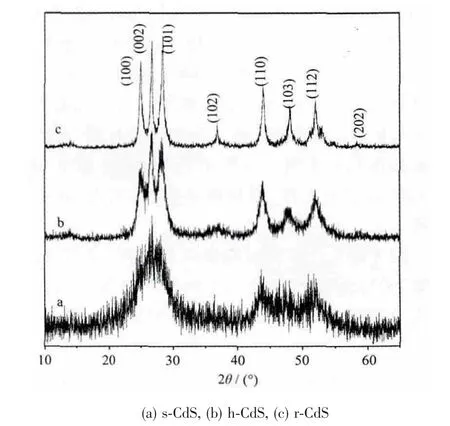
图1 CdS 的X-射线衍射图Fig.1 XRD patterns of CdS

图2 实心球透射电镜照片(a), 实心球透射电镜放大照片(b),实心球选区电子衍射图(c),实心球高分辨电镜照片(d),实心球扫描电镜照片(e)Fig.2 TEM image (a), the amplificatory image (b), SAED pattern (c), HRTEM image (d), and FESEM image (e) of solid CdS nanospheres. The samples were obtained after reaction with GSH as the sulfur source at 120 ℃for 24 h
其中,图2b 为透射电镜的放大照片,可以看出球体中间为实心,而且边缘包裹一层薄膜状物,可能是没有反应掉的GSH。图2d 为高分辨透射照片,可以看出组成实心球边缘的颗粒是结晶度不高(没有看到格栅)的CdS 小颗粒无序生长而成。通过其衍射环(图2c)可以进一步确认120 ℃制备的CdS 样品为六方相纤维锌矿型。对于多晶化合物,经电子衍射,若结晶度高则出现明显的衍射环; 若结晶差则衍射环为光晕环。通过选区电子衍射SAED 图,可以明显地看出各晶面在同一点打出了光晕环,故表明实心球CdS 为结构无序,结晶度不高,这与XRD 的分析结果一致。
图3 是h-CdS 样品电镜照片组图。从图3a 可以得知,水热温度控制在160 ℃条件制备出的CdS 样品为中间区域较白,而边缘比较黑的球状形貌,这一特征与文献报道的空心球结构基本一致。球的直径主要集中在100~200 nm,外壳厚大约35 nm。通过图3e 可以看出,箭头所指部分为球体在制备过程中自然破裂或者塌陷,进一步证明160 ℃条件下制备出的CdS 纳米球为空心。为了进一步了解空心球表面的结构,图3b 给出放大的透射电镜照片,可以看出空心球外壳是由粒径约8 nm 左右的亚微晶颗粒有序生长并堆积而成。通过高分辨电镜照片(图3d)可以看出六方相纤维锌矿CdS 晶格间距为0.320 和0.340 nm 对应的(101)和(002)晶面,而且通过选区电子衍射SAED 图(图3c),可以明显地看出各晶面在同一点打出了明显的光环,表明h-CdS 为六方相纤维锌矿型,结构有序,结晶完好,与XRD 的分析结果一致。
图4 为r-CdS 样品电镜照片组图。从图4a 可以看出水热温度控制在200 ℃下制备的CdS 样品90%以上的微观形貌为棒状外形,纳米棒宽约为20~100 nm,长约为800~2 000 nm。从图4e 还可以看出制备的样品中还存在少量的颗粒和球状CdS。这一现象也为研究CdS 不同形貌的生长机理提供一些依据。图4b 为放大的透射电镜照片,可以明显看出六方相CdS 的棱,同时高分辨电镜照片(图4d)清晰的看见晶面间距(d=0.34 nm)对应的(002)晶面,可以证明CdS 纳米棒是沿c 轴取向排列生长而成。通过选区电子衍射(图4c),也可以看出各晶面在同一点打出了明显的衍射光斑,显示纳米棒为单晶结构,表明r-CdS 为六方相纤维锌矿型,结构有序,结晶完好,这与XRD 的分析结果也很吻合。

图3 空心球透射电镜照片(a), 空心球透射电镜放大照片(b),空心球选区电子衍射图(c),实心球高分辨电镜照片(d),实心球扫描电镜照片(e)Fig.3 TEM image (a), the amplificatory image (b), SAED pattern (c), HRTEM image (d), and FESEM image (e)of hollow CdS nanospheres.The samples were obtained after reaction with GSH as the sulfur source at 160 ℃for 24 h
图5 为不同形貌样品微区成分的X 射线能谱仪(EDS)分析图,结果表明不同水热温度下制备的CdS 表面Cd2+和S2-含量不同,不同形貌的s-CdS、h-CdS 和r-CdS 对 应 的nS2-/nCd2+比 分 别 为1.03、1.01 和0.98。其中,Cu 和C 分别来源于铜网和碳膜。另外,在图5a、5b 和5c 都发现O 存在,而且随着制备温度提高O 含量降低。可以解释为S 和O 来源于CdS表面过量的没有反应掉的GSH,此结果也进一步证实水热温度的提高,会加速GSH 中S-C 键断裂和分解,结构导向剂GSH 的分解快慢对微观形貌的形成具有很大的影响。不同形貌CdS 表面的nS2-/nCd2+比不同,也可能来自于晶体表面的缺陷,这一现象对不同形貌样品的光解水产氢性能存在一定的影响。
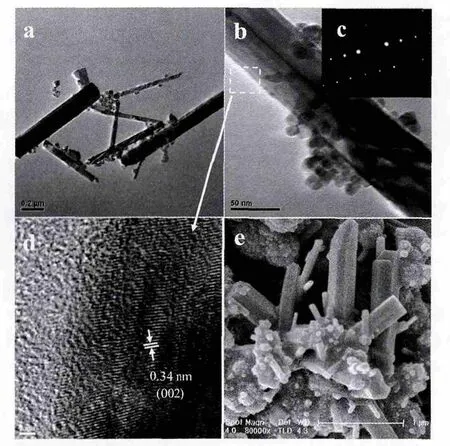
图4 纳米棒透射电镜照片(a), 纳米棒透射电镜放大照片(b),纳米棒选区电子衍射图(c),纳米棒高分辨电镜照片(d),纳米棒扫描电镜照片(e)Fig.4 TEM image (a), the amplificatory image (b), SAED pattern (c), HRTEM image (d), and FESEM image (e) of CdS nanorods.The samples were obtained after reaction with GSH as the sulfur source at 200 ℃for 24 h
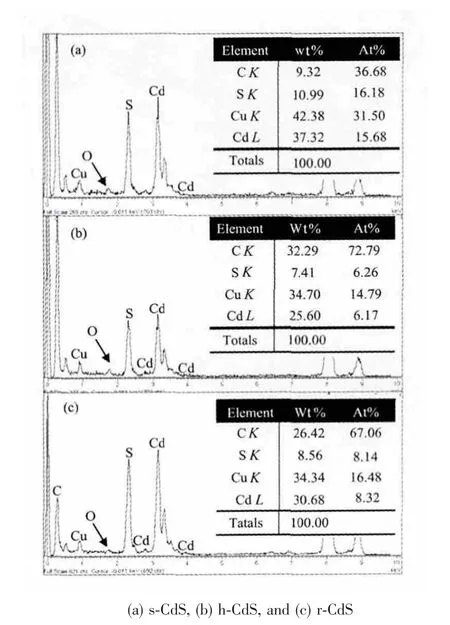
图5 EDS 谱图Fig.5 EDS spectrum
2.3 s-CdS、h-CdS 和r-CdS 紫外可见吸收光谱图
催化剂的光学吸收性能对其光催化能力具有较大的影响,图6 为s-CdS、h-CdS 和r-CdS 的紫外可见光吸收光谱。由图可知,s-CdS 的吸收边在465 nm,而h-CdS 和r-CdS 吸收边分别在515 和531 nm。表明制备的CdS 样品具有很好的响应可见光能力。3 种微观形貌的CdS 带宽分别为2.67、2.41 eV和2.34 eV。由于CdS 属于直接跃迁半导体,当其颗粒尺寸减小到纳米级时(即粒径在1~100 nm,粒径与德布罗意波长相近时),即可显示出明显的“量子尺寸效应”。在紫外可见光吸收光谱图上表现为吸收边往短波长移动(蓝移)。结合电镜照片,s-CdS 和h-CdS 的球体直径基本上相差不大,而在吸收边却相差50 nm,可归结于组成纳米球的亚微粒子粒径差异而致,组成实心球的粒径非常小,发生了量子尺寸效应。然而,r-CdS 由于粒径比较大,基本接近体相材料,吸收边和体相材料也比较相近。在光催化反应过程中真正起到光作用的是表面粒子,故表面粒子结构很大的影响了材料的宏观性能。

图6 紫外可见光吸收光谱图Fig.6 UV-visible diffuse reflectance spectra
2.4 s-CdS、h-CdS 和r-CdS 荧光光谱图
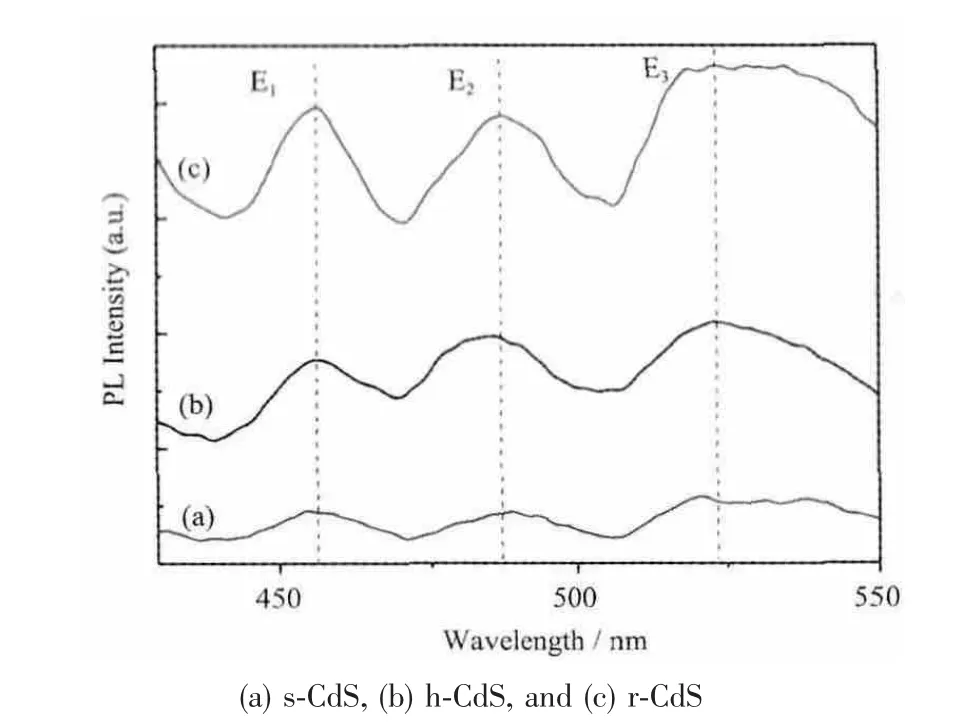
图7 荧光光谱图Fig.7 PL spectra of samples
从图7 可以看出s-CdS、h-CdS 和r-CdS 有相似的荧光发射光谱图,当用346 nm 波长光激发时,它们都在455 nm(紫光)、487 nm(蓝光)和523 nm(绿光)出现3 个宽的发射光谱。通常高能E1区荧光发射峰归结为电子空穴对复合引起的近带发射光谱;而E2和E3可以归结为电子给体的激发或者(和)电子空穴对的浅层捕获引起的发射光谱,这类荧光发射光谱通常暗示CdS 晶体的某些内部特征或者表面缺陷,特别是来自颗粒表面缺陷[15]。这一结果同EDS 显示的s-CdS、h-CdS 和r-CdS 表面存在过量S 或者Cd一致。相同的荧光现象在CdS 空心纳米链[16]、微笼状[13]、空心球[17]、纳米带和薄盘[18]及纳米棒[19]也有报道。众所周知,荧光强度与电子/空穴对复合有直接关联,荧光越强说明电子/空穴对复合几率越大,从而导致该催化剂的光催化性能降低。从图7 可以看出3 种CdS 荧光强度顺序为s-CdS<h-CdS<r-CdS。这一结论与后面的光催化分解水产氢活性一致。另外,由晶体缺陷造成的荧光光谱没有出现红移,说明制备的CdS 样品有很好的光学属性。但是,该类的荧光强度越强说明表面缺陷越多,也导致催化活性的降低。
2.5 s-CdS、h-CdS 和r-CdS 等温吸脱附曲线与孔径分布图

图8 等温吸脱附曲线图Fig.8 Nitrogen adsorption/desorption isotherm

图9 BJH 孔径分布Fig.9 Poresize distribution (BJH)
图8 为s-CdS、h-CdS 和r-CdS 的 氮 气 在77 K下的等温吸脱附曲线图。由图可知,s-CdS、h-CdS 和r-CdS 曲线均属于Ⅱ型,显示材料为无孔或者大孔的微观结构,主要吸附特征为单层-多层吸附。对氮气吸附能力为h-CdS>r-CdS>s-CdS,且属于H3 型滞后环,可以归结为颗粒堆积形成的狭缝型孔结构,并且在较高相对压力时吸附量仍在不断增加。图9 为孔径分布图。结果显示h-CdS 平均孔径分布主要在3.8 nm;s-CdS 主要集中在3.5 nm; 而r-CdS 平均孔径分布主要在3.8 nm 和10~20 nm 这两个区域内,可能原因是制备纳米棒时出现少量的纳米颗粒和纳米球堆积导致。通常孔径分布在小于10 nm 以内认为是纳米球外壁上的颗粒堆积造成的,而大于10 nm 可以归为纳米棒堆积造成的,相同的现象在钛酸盐纳米管中也有报道[20]。这一结论与电镜照片提供的信息一致,进一步证明制备的实心球和空心球是由许多亚微晶堆积而成; 实心球的滞后环要比空心球的滞后环往高的相对压力移动,说明堆积比较紧密,组成实心球的亚微晶粒比较小;而空心球的滞后环比较大也往低的相对压力移动,说明空心球的堆积孔来源于粒径相对比较大的CdS 颗粒堆积而成。表1 列出了s-CdS、h-CdS 和r-CdS 的BET 比表面积和孔参数。

表1 s-CdS,h-Cd 和r-CdS 的BET 比表面积和孔参数Table 1 BETsurface areas and pore parameters of s-CdS, h-CdS and r-CdS
2.6 s-CdS、h-CdS 和r-CdS 可见光分解水产氢活性测试
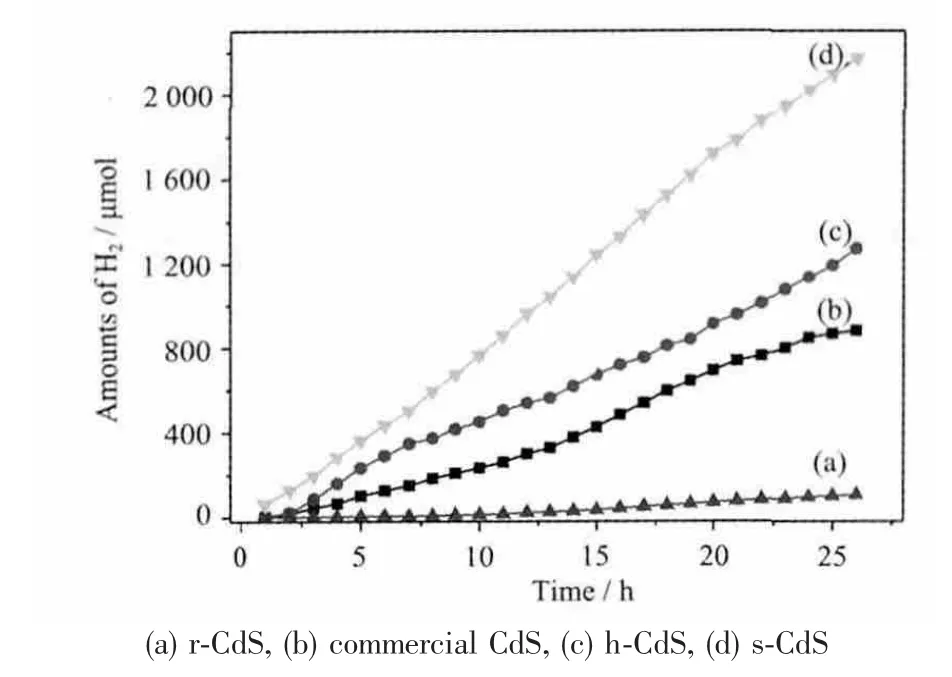
图10 产氢活性随时间的变化曲线Fig.10 Amount of H2 evolved vs irradiation time
在可见光下(λ≥420 nm),s-CdS、h-CdS 和r-CdS与商业CdS 对比的分解水产氢量随时间的变化曲线见图10。s-CdS 表现出最高的产生氢气能力,25 h累计可达2 100 μmol,而且在25 h 内催化剂相对比较稳定;h-CdS 氢产率在开始比s-CdS 稍低点,当反应进行到7 h 后快速降低,25 h 累计产生氢气量是s-CdS 一半。r-CdS 活性最差,甚至比商业CdS 还要低,相同时间内s-CdS 氢气产量比其高2 个数量级。以上产氢结果可以说明不同形貌的CdS 光解水产氢性能相差很大,可以得出结论:催化剂制备方法和表观的形貌结构在光分解水产氢过程中起到很大的作用。不同形貌的CdS 光解水产氢性能差异的主要原因可以解释如下: 光催化作用主要发生在催化剂的表面,当催化剂受到光照射时,电子空穴对发生激发并迁移到表面与溶液反应,生成氢气。电子-空穴对的迁移越快,就会抑制它们的复合,相应的氢气产率就会大大的提高。具体到实心球、空心球和棒状微观形貌,虽然s-CdS 和h-CdS 表面都是由亚微晶颗粒组成,光催化分解水产氢都发生在这些亚微晶表面上;但从电镜照片以及由于“量子尺寸效应”引起s-CdS 紫外可见光吸收边蓝移现象,可以推测s-CdS的表面亚微晶的相对比较小的粒径在光催化过程中起到决定性作用。从荧光强度以及EDS 给出的表面缺陷也可以进一步指出s-CdS 表面亚微晶产生的电子-空穴复合强度要低于其它形貌的材料。对于r-CdS 而言,小的比表面积可能是造成产氢活性比较低的主要原因。
3 结 论
以还原型谷胱甘肽作为硫源和结构导向剂 “一壶”水热法制备系列CdS 光催化材料。通过调节反应物质的nCd/nS比和水热温度等参数可控的制备出分散性好的CdS 实心纳米球、空心纳米球以及纳米棒等不同微观形貌结构的光催化材料。对比研究了不同形貌光催化剂的光解水产氢的宏观性能,发现s-CdS 产氢活性最高,h-CdS 次之,r-CdS 最差。这一结果可归结于实心球表面亚微晶的粒径相比其它形貌的小,导致电子-空穴对快速迁移至表面并与溶液反应,抑制体相复合,导致生成的氢气量大大的提高。针对本文报道的3 种形貌结构材料,可以得出结论: 结构相对比较简单的实心球在光催化过程中活性要优于微观形貌相对比较复杂物质的构效关系规律。
[1] Fujishima A, Honda K. Nature, 1972,238(5358):37-38
[2] Li C L, Yuan J, Shangguan W F, et al. Inte. J. Hydrogen Energy, 2010,35:7073-7079
[3] LI Cao-Long(李曹龙), CHEN Wei(陈威), YUAN Jian(袁坚),et al. Acta Phys.-Chim. Sin. (Wuli Huaxue Xuebao), 2012,28(02):450-456
[4] CHEN Wei(陈威), GAO Han-Yang(高寒阳), YANG Yu(杨 宇), et al. Acta Phys.-Chim. Sin. (Wuli Huaxue Xuebao),2012,28(12):2911-2916
[5] Zong X, Han J F, Ma G J, et al. J. Phys. Chem. C, 2011,115:12202-12208
[6] LIN Pei-Bin(林培宾), YANG Yu(杨宇), CHEN Wei(陈威),et al. Acta Phys.-Chim. Sin. (Wuli Huaxue Xuebao), 2013,29 (6):1313-1318
[7] Bao N Z, Shen L M, Domen K, et al. Chem. Lett., 2006,35:318-327
[8] Bao N Z, Shen L M, Takata T, et al. Chem. Mater., 2008,20:110-117
[9] Luo M, Liu Y, Hu J C, et al. Appl. Mater. Interfaces,2012,4:18131821
[10]ZHANG Qin-Feng(张钦峰), HUANG Jian-Feng(黄剑锋),CAO Li-Yun(曹 丽 云), et al. Chinese J. Inorg. Chem.(Wuji Huaxue Xuebao), 2013,29(2):271-276
[11]Ke D N, Liu S L, Dai K, et al. J. Phys. Chem. C, 2009,113:1602116026
[12]Girginer B, Galli G, Chiellini E, et al. Inte. J. Hydrogen Energy, 2009,34:1176-1184
[13]Gong Q, Qin X F, Zhou P L, et al. J. Phys. Chem. C, 2007,111:193540-193551
[14]GAO Jian( 高 洁), LONG Fei ( 龙 飞), CHI Shang-Sen(池 上 森), et al. Chinese J. Inorg. Chem.(Wuji Huaxue Xuebao), 2012,28(8):1656-1600
[15]Nanda K K,Kruis F E,Fissan H.Nano Lett.,2001,1:605-616
[16]Liu J K, Luo C X, Yang X H, et al. Mater. Lett., 2009,63:124-130
[17]Dai Z H, Zhang J, Bao J C, et al. J. Mater. Chem., 2007,17:1087-1093
[18]Li C L, Yuan J, Han B Y, et al. Inte. J. Hydrogen Energy,2011,36:4271-4279
[19]Cai W, Li Z G, Sui J H, et al. Nanotechnology, 2008,19:465606-465616
[20]Lee C K, Wang C C, Lyu M D, et al. J. Colloid Interface Sci., 2007,316:562-569
