无需逆转录的高通量多重呼吸道病毒RNA检测技术的建立
2013-08-09杨丹丽田晓怡石伟先
杨丹丽,田晓怡,石伟先,郑 直
1中国医学科学院 北京协和医学院 基础医学研究所生物化学与分子生物学系,北京 100005
2北京市疾病预防控制中心传染病地方病控制所,北京 100013
呼吸道病毒是最常见的呼吸道病原体之一,能引起人群严重的呼吸道感染,具有较高的发病率和死亡率。据报道A和B型流感病毒、呼吸道合胞病毒、1、2和3型副流感病毒是婴幼儿呼吸道感染的最主要诱因[1]。近几年报道的冠状病毒、鼻病毒、偏肺病毒等也是引起人类呼吸道感染的重要病毒[2-4]。呼吸道病毒暴发性强,如SARS、禽流感等病毒的暴发,给人类社会和国民经济带来很大负担[5]。由于临床上待检测样品数量多,且待测呼吸道病毒种类繁多,变异性强,这给临床诊断、治疗及疾病监控带来很大困难。现有方法如直接荧光抗体、细胞培养及各种PCR方法等检测方法在灵敏度、特异性及样品通量方面存在较大局限性,这使得对呼吸道病毒难以实现大规模、高通量的检测筛查和监测。本研究直接以病毒RNA为检测靶标,无需逆转录、靶标扩增等样品前处理过程,利用以微球为基础的高通量平台,采取本课题组以往成功开发并应用的RNA夹层杂交反应捕获靶标RNA[6-7],然后通过分支DNA技术[8]放大信号,对待测样本中存在的呼吸道病毒进行定性定量分析。
材料和方法
病毒总RNA 选用北京市疾病预防控制中心冻存的、经实验室PCR确诊感染8种病毒之一、且病毒含量较高的咽拭子样本200 μl,使用QIAamp Viral RNA Mini Kit(QIAGEN公司)提取病毒总RNA。
靶标病毒体外转录RNA 以各病毒的外转录RNA(in vitro-transcribed RNA,IVT-RNA)作为检测靶标。从NCBI下载病毒的靶标核酸序列 (毒株序列号见表1),并查到各病毒的属内保守区。利用Primer Premier 5.0软件设计位于保守区内的病毒特异性逆转录引物和PCR扩增引物 (所有引物由Invitrogen公司合成,用PAGE法纯化),对病毒总RNA进行特异逆转录及cDNA的特异扩增。按照标准克隆流程获得含目的片段的质粒并测序正确后,对质粒进行体外转录和纯化 (Life Technology MEGAscript转录试剂盒),获得体外转录靶标RNA即IVT-RNA。
病毒RNA检测技术、病毒靶标及探针设计 利用本课题组以往研究的RNA夹层杂交结合信号放大技术和Luminex微球技术[6-7]建立一个多重检测8种病毒RNA的方法。该方法通过在不同颜色的Luminex微球表面上进行一系列的核酸片段杂交来完成检测。检测用寡核苷酸探针包含有与靶标RNA特异性配对的序列,以及额外的3’端“尾巴”序列用于和微球表面或信号检测系统特异性连接。这些探针分别称作捕获探针 (capture extender,CE)和检测探针 (label extender,LE)[6-7]。CE探针的一部分与偶联于微球表面的捕捉序列结合,另一部分与靶标序列结合,杂交之后,就可以将靶标序列固定下来。信号的放大是通过DNA信号放大分子介导的杂交进行的:LE探针的一部分与靶标序列结合,其尾部与DNA信号放大分子结合,最终通过荧光检测DNA信号放大分子的存在及数量。
本研究作为对方法学的可行性研究和初步表征,采用体外合成的靶标病毒RNA(IVT-RNA)为检测对象,将其加入咽拭子采集液的裂解液中,以模拟病毒直接裂解所释放的RNA。对于A和B型流感病毒、B型呼吸道合胞病毒、1和2型副流感病毒等5种待测病毒,选取了GenBank中该病毒某一特定毒株,以基因组中针对该病毒特异的病毒基因片段序列作为靶标,选择不同毒株间变异程度较低的保守区域设计检测探针 (表1)以便检测不同毒株。由于用于合成IVT-RNA的质粒由临床样品克隆所得,其序列和设计探针时采用的GenBank序列并不完全相同,因此探针序列和待测靶标IVT-RNA序列不完全互补 (表1)。对于A型呼吸道合胞病毒,3型副流感病毒和偏肺病毒,探针则根据IVT-RNA设计,序列完全互补。
本技术工作流程类似于ELISA,可以在不进行逆转录和靶标扩增的情况下,同时进行8种RNA检测。可一次实验并行检测96个样品。
病毒RNA检测方法 针对每一种病毒靶标RNA,本研究按已发表的设计原理[9],经过实验优化,设计了一套能与Luminex检测系统兼容的寡核苷酸探针。表1列出了每种病毒的探针数目。探针以外所需检测试剂由Quantigene Plex 2.0 Assay试剂盒提供 (Panomics公司)。将1 μl包含8种病毒的IVTRNA混合物与32.4 μl咽拭子采集液、66.6 μl裂解液、8 μl 8种病毒探针混合物、以及2 μl磁性微球混合物 (包含8种已包被不同捕获探针的磁性微球)混合后转移至1个96孔板的孔中,密封后置于54℃空气浴摇床 (Vortemp 56型,Labnet公司)孵育40 h,然后将96孔板置于磁铁上,磁性微球吸附在孔底部后,将溶液 (包含未杂交的探针等)弃去,加入洗涤液对微球进行洗涤后,根据试剂盒说明书要求对微球上的靶标分子按步骤进行荧光信号放大,最终通过Bio-Plex 200荧光微球检测仪 (BioRad公司)分析微球的荧光信号[6-7]。
特异性检测 为了检查是否针对某种病毒的探针会和其他病毒非特异性结合产生交叉反应信号,在反应体系中加入针对8种病毒的微球及探针混合物,但只加一种病毒的IVT-RNA(0.5 amol/μl,相当于3.01×105个拷贝),检测每种微球上的信号值。通过比较非靶标病毒微球荧光信号值与背景荧光信号值,评估检测系统的特异性。采用t检验进行统计学分析。
灵敏度检测 IVT-RNA经过A260浓度定量 (Nanodrop 2000)后,用0.1 g/L酵母tRNA(Life Technology)对其进行4倍系列浓度稀释,最终获得8至0.002 amol/L的7个浓度的一系列IVT-RNA。通过检测该系列已知浓度样品的信号值,建立IVT-RNA的标准曲线。检测灵敏度即检测下限设定为:标准曲线的低端开始偏离线性时的拷贝数值。
结 果
检测特异性 在只有一种病毒RNA存在时,仅靶标病毒微球产生特异性荧光信号。对于其他病毒微球,在高达3.01×105个 (0.5 amol/μl)非特异性RNA分子存在情况下,产生的非特异信号与空白对照的差异无统计学意义 (P均>0.2,图1)。
检测灵敏度和线性范围 标准曲线显示:此检测方法对8种病毒IVT-RNA的检测灵敏度在0.002~0.0078 amol之间,也即1204~4695个拷贝的RNA分子,具有较高的灵敏度。本方法的线性范围在0.008~2 amol(图2)。
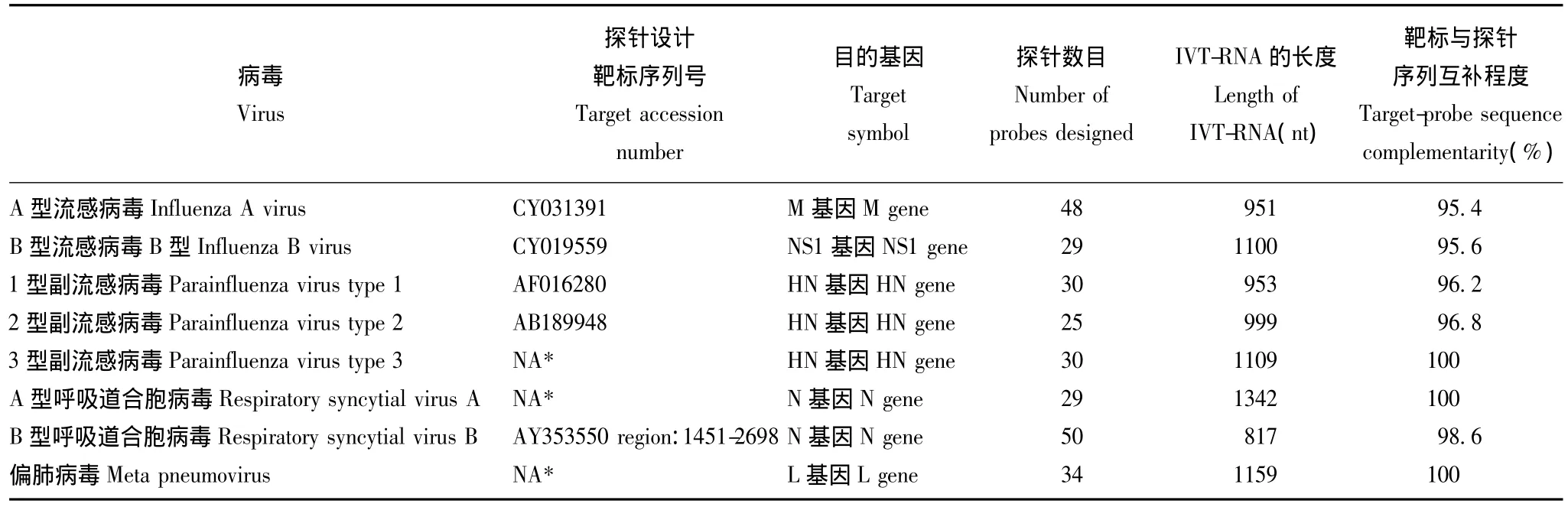
表1 靶标呼吸道病毒的信息Table 1 Information of the respiratory virus targets and probes
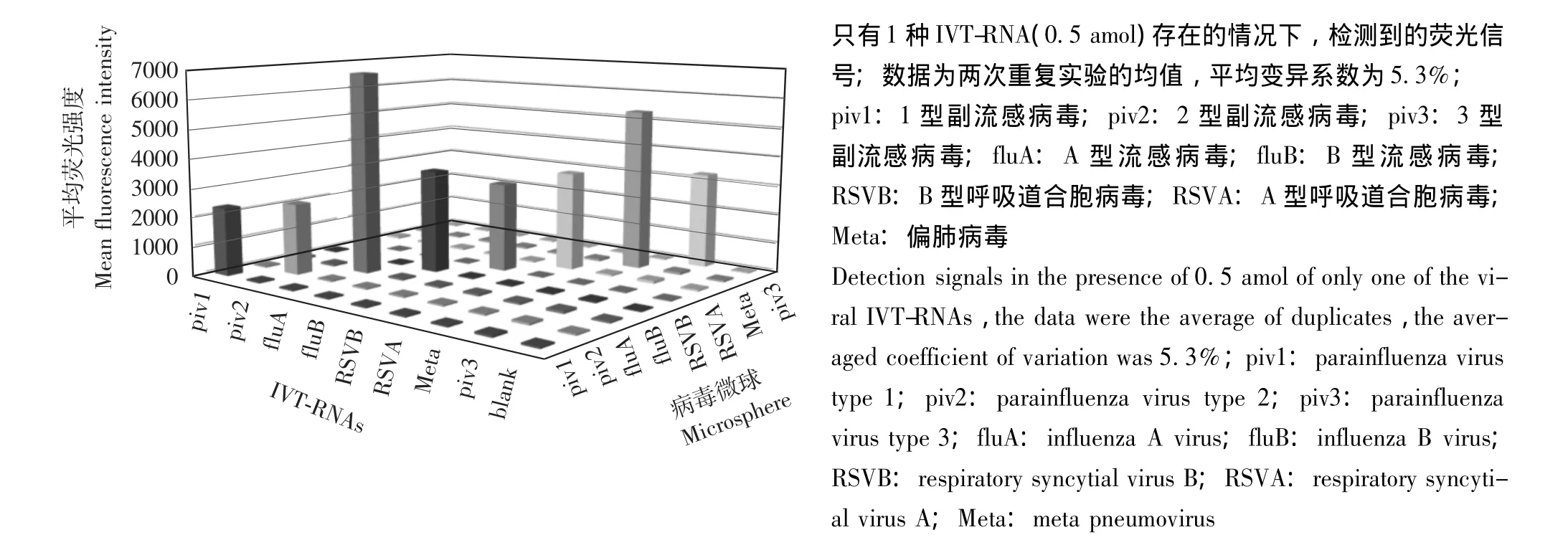
图1 高通量核酸检测系统的特异性评估Fig 1 Specificity of the high-throughput respiratory virus detection method
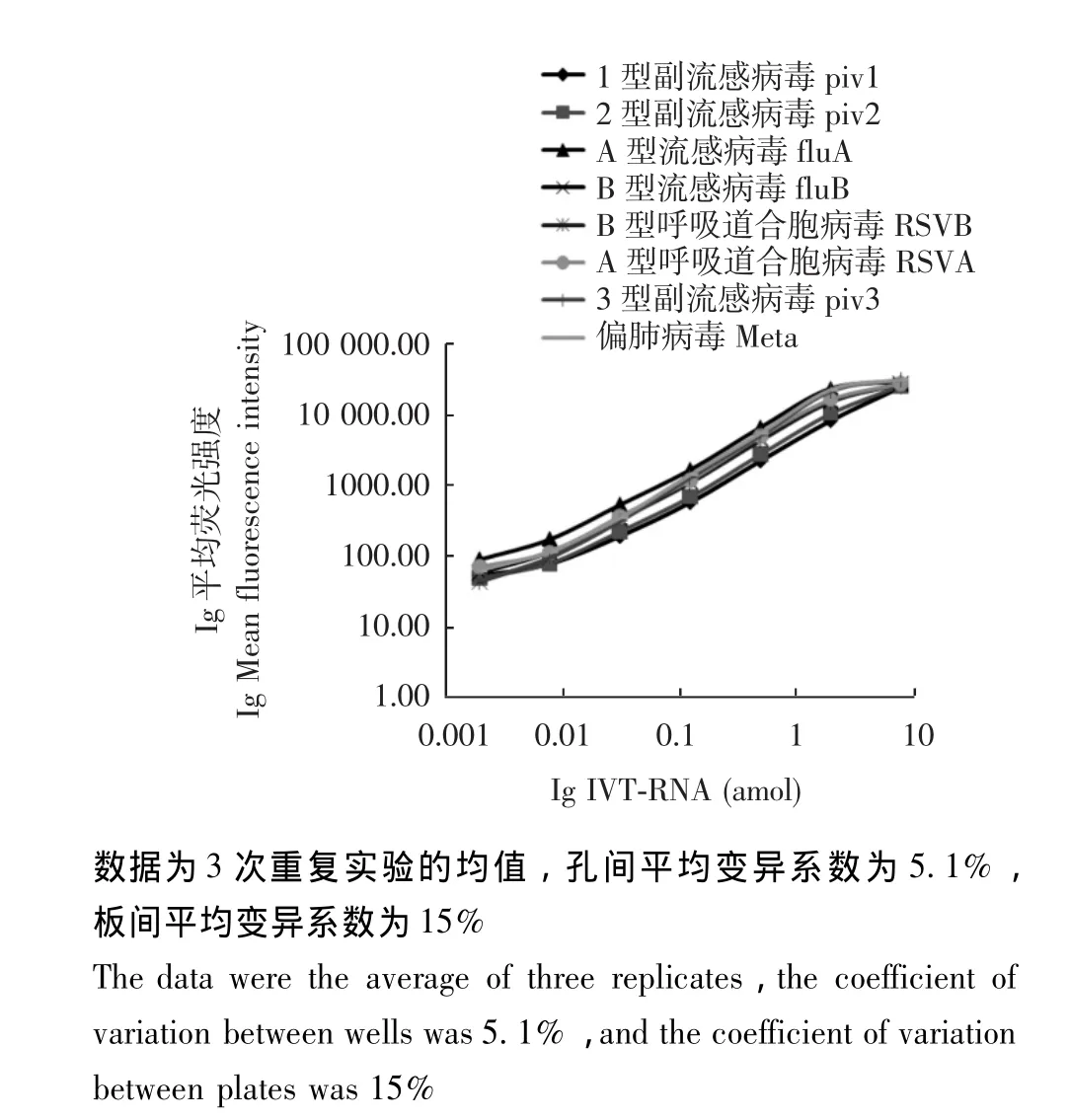
图2 梯度稀释的IVT-RNA评估高通量检测方法的灵敏度Fig 2 Sensitivity assessment using serially diluted IVT-RNAs for all 8 viruses
讨 论
急性呼吸道感染绝大多数由病毒引起。由于这些病毒感染导致的临床表现很相似,很难根据临床症状准确判断致病原。据报道,在住院儿童中,仍然有20% ~30%的病原体还无法明确诊断[10]。在不同季节、不同年龄段流行的主要病毒也不同[11]。为了确切诊断出致病原,临床病毒学实验室通常使用病毒培养及血清学方法来检测常见呼吸道病毒。病毒的细胞培养是诊断的金标准,但是细胞培养时间长,劳动强度大,不利于病毒的快速诊断;血清学方法快速,在较短的时间内就可以获得检测结果[12],但是它往往需要病毒感染人体一段时间后才能检测,并且需要已知的特定抗原或抗体,目前现有抗原抗体通常不能满足诊断要求[13]。
近些年来,分子诊断技术被陆续用于呼吸道病毒 检 测,如 RT-PCR[14]、real-time PCR[15]等。这 些诊断技术不仅灵敏度高,特异性也相当好。传统的RT-PCR方法一次只能从一个标本中检出一种病毒。最近王春等[12]用多重RT-PCR方法弥补了这一缺陷,但PCR后续检测流程依靠电泳,应用于大样本量的研究有很大的局限性。Mahoney等[16]用多重PCR扩增不同类型的病毒分子,结合xMAP多重分析平台检测信号,简化了后续检测过程,得到了较高的检测灵敏度和检测通量。但是以上的多重PCR检测技术都需要前期样品处理,包括对核酸进行逆转录,标记或扩增等,重复性差。有证据表明:由不同实验室使用RT-PCR这一技术检测同一样品,往往会得出差异较大的结果[17];其次,对样品复杂的前处理过程极大地限制了此类检测方法的通量,使得在大规模监测时难以应用。另外,由于呼吸道病毒的变异性较高,引物结合处的序列变异将造成PCR反应失败形成假阴性结果。最后,PCR反应的污染问题也是一大难题,很容易出现微量污染造成的假阳性结果,这就限制了多重PCR类方法在普通临床实验室的应用[18]。总之,目前呼吸道病毒检测方法无法同时满足临床上对灵敏度、特异性和高通量的要求。
本研究直接以病毒RNA作为检测靶标,无需逆转录、靶标扩增等常规样品前处理步骤,不仅简化了操作步骤,还避免了样品处理过程中可能存在的差异,使得误差显著降低 (平均变异系数约5%)。本方法一次实验可同时检测96个样品,对每一个样品可同时进行8种常见病毒的检测,所需样本量很少。本研究数据表明该方法具有很好的特异性和较高的灵敏度,显示此方法用于检测常见呼吸道病毒有较高的的可行性。尽管从样品裂解到得出结果需要3 d的时间,但人工操作时间仅2~3 h。对于需要连续检测大批量样品的检测筛查,本方法能够克服目前检测方法在通量上的局限性,节约检测成本,提高检测通量。另外,此检测系统针对每一靶标RNA都至少有20条寡核苷酸探针,即使病毒在保守区存在约5%的变异,本方法仍可以获得很好的结果。最后,与PCR直接放大检测靶标不同,本方法采用放大检测信号的策略,可以避免由于产物交叉污染造成的假阳性结果。
综上,本方法用于高通量多种呼吸道病毒检测,同现有方法相比具有较明显的优势,特别适用于流行病学监测监控及传染病大规模筛查。该课题组下一步将进行高通量流行病临床标本的检测,以评估该方法在传染病监测预防中的作用。
[1]Hall CB.Respiratory syncytial virus and parainfluenza virus[J].N Engl J Med,2001,344(25):1917-1928.
[2]Gaunt ER,Hardie A,Claas EC,et al.Epidemiology and clinical presentations of the four human coronaviruses 229E,HKU1,NL63,and OC43 detected over 3 years using a novel multiplex real-time PCR method [J].J Clin Microbiol,2010,48(8):2940-2947.
[3]Jackson DJ.The role of rhinovirus infections in the development of early childhood asthma[J].Curr Opin Allergy Clin Immunol,2010,10(2):133-138.
[4]Broor S,Bharaj P,Chahar HS.Human metapneumovirus:a new respiratory pathogen[J].J Biosci,2008,33(4):483-493.
[5]Cunha BA.The atypical pneumonias:clinical diagnosis and importance[J].Clin Microbiol Infect,2006,12(Suppl 3):12-24.
[6]Flagella M,Bui S,Zheng Z,et al.A multiplex branched DNA assay for parallel quantitative gene expression profiling[J].Anal Biochem,2006,352(1):50-60.
[7]Zheng Z,Luo Y,McMaster GK.Sensitive and quantitative measurement of gene expression directly from a small amount of whole blood[J].Clin Chem,2006,52(7):1294-1302.
[8]Urdea MS.Branched DNA signal amplification [J].Biotechnology(N Y),1994,12(9):926-928.
[9]Bushnell S,Budde J,Catino T,et al.Probe designer:for the design of probesets for branched DNA(bDNA)signal amplification assays[J].Bioinformatics,1999,15(5):348-355.
[10]Freymuth F,Quibriac M,Petitjean J,et al.Viruses responsible for respiratory infections in pediatrics.Evaluation of 3,480 nasal aspirates performed in children over a 6-year period[J].Ann Pediatr(Paris),1987,34(7):493-501.
[11]黄芳,石伟先,崔淑娟,等.北京地区2010年10月至2011年4月急性呼吸道感染病毒流行特征分析[J].国际病毒学杂志,2011,18(4):98-100.
[12]王春,赵百慧,张泓,等.上海市儿童下呼吸道感染常见病毒诊断方法比较[J].检验医学,2011,26(9):589-592.
[13]Yan Y,Zhang S,Tang YW.Molecular assays for the detection and characterization of respiratory viruses[J].Semin Respir Crit Care Med,2011,32(4):512-526.
[14]Suarez P,Zardoya R,Prieto C,et al.Direct detection of the porcine reproductive and respiratory syndrome(PRRS)virus by reverse polymerase chain reaction(RT-PCR)[J].Arch Virol,1994,135(1-2):89-99.
[15]Kaltenboeck B,Wang C.Advances in real-time PCR:application to clinical laboratory diagnostics [J].Adv Clin Chem,2005,40:219-259.
[16]Mahoney J,Chong S,Merante F,et al.Development of a respiratory virus panel test for detection of twenty human respiratory viruses by use of multiplex PCR and a fluid microbead-based assay[J].J Clin Microbiol,2007,45(9):2965-2970.
[17]Ramsden SC,Daly S,Geilenkeuser WJ,et al.EQUAL-quant:an international external quality assessment scheme for real-time PCR [J].Clin Chem,2006,52(8):1584-1591.
[18]Mandy FF,Nakamura T,Bergeron M,et al.Overview and application of suspension array technology[J].Clin Lab Med,2001,21(4):713-729,vii.
