基于多级质谱的蛋白质组定量新方法新技术进展
2013-07-13张莹杨芃原陆豪杰
张莹,杨芃原,陆豪杰
(复旦大学化学系和生物医学研究院,上海 200032)
随着蛋白质组学研究的深入和发展,大量功能蛋白质和疾病相关的潜在蛋白质标志物被发现和鉴定。早期蛋白质组学研究比较重视鉴定蛋白质和蛋白质修饰信息,而越来越多的研究表明,疾病或不同生理状态相关的重要蛋白质往往不仅是种类的改变,更多的是蛋白质量的改变。因此,从定性走向定量已经成为蛋白质组研究的必然趋势[1]。
生物质谱作为蛋白质组研究的核心技术,不仅承担着蛋白质鉴定的任务,在蛋白质定量中也扮演着更为重要的角色。随着定量蛋白质组学研究的不断发展,越来越多的基于生物质谱的蛋白质组定量方法应运而生。从蛋白质组定量所要达到的目的上大致可以分为绝对定量和相对定量两大类;从蛋白质定量所采取的策略上可根据标记的有无分为非标记定量和标记定量两大类;在标记定量中普遍采用的是稳定同位素标记技术,可以根据标记方法细分到体内代谢标记、酶促标记和化学标记;还可以根据质谱定量信息来源的不同分为一级质谱(MS1)定量和多级质谱(MSn)定量。
多级质谱定量,顾名思义,其定量信息来自多级质谱谱图。通常是首先在蛋白质酶解产生的肽段上引入稳定同位素标记,然后比较在多级质谱中带有同位素标记的报告离子或带有同位素标记的肽段碎片离子的质谱峰强度来进行蛋白质的相对定量。根据肽段的不同标记方法、实现质谱碎裂的不同方式以及定量的不同信息,二级质谱定量方法又可以再分为以下几种:开大窗口二级质谱定量,等重标记结合报告离子二级质谱定量及相应的利用互补离子的二级质谱定量,等重标记结合b、y 离子对的二级质谱定量。多级质谱定量和一级质谱定量相比,由于经过了肽段母离子选择,大大降低了二级质谱谱图中的噪声水平,从而提高了二级质谱谱图的信噪比,进而可以提高肽段和蛋白质定量的准确性。本文将重点综述基于多级质谱的蛋白质组定量新方法、新技术及它们的应用。
1 开大母离子选择窗口二级质谱定量
开大母离子选择窗口实现二级质谱定量是一种比较早期的二级质谱定量方法,常见的适合于一级质谱定量的稳定同位素标记方法理论上都可通过开大窗口实现二级质谱定量。通过开大母离子的质量选择窗口(一般为10 Da),将一级质谱中轻、重同位素标记的肽段同时选择进行共碎裂,从而在二级质谱中得到轻、重标记的成对碎片离子对,用这些碎片离子对来进行肽段的定量。基于代谢标记[2,3]和18O标记[4]均可以通过开大母离子选择窗口在二级质谱中实现定量。此类方法具有的优势如下:首先,该方法只需调节母离子的选择窗口即可实现二级质谱定量,不会额外增加样品的预处理步骤,在操作上易于实现;第二,信噪比较高的二级谱图有利于提高定量的灵敏度;第三,谱图中多对碎片离子用于定量有效地降低了肽段定量的随机误差,在一定程度上提高了定量的准确性。但通过开大窗口来实现二级质谱定量也有很多缺陷导致它不能在复杂蛋白质样品中最终被广泛应用,除了因为轻、重标记的肽段增加了一级谱图的复杂性,最主要的是在二级质谱中,放大的母离子选择窗口增加了色谱共洗脱质量数相近的干扰肽段发生共碎裂的几率,这些对肽段的鉴定可靠性和定量准确性均有影响。
2 等重标记二级质谱定量
等重标记则很好地避免了上述开大窗口定量方法中增加一级谱图复杂性的缺点,特别是避免了开大窗口导致色谱共洗脱质量数相近肽段的干扰问题,提高了定量的灵敏度和准确度。等重标记的二级质谱定量方法可根据定量离子的来源分为单个离子和多对离子定量的方法,其中多对离子定量方法在很大程度上消除了单个离子定量时的不准确性。
2.1 基于单个离子定量
2.1.1 基于低质量端报告离子定量
基于单个离子定量的方法中,最具有代表性的是基于商业化等重同位素标记试剂iTRAQ(isobaric tag for relative and absolute quantitation)[5]和TMT(tandem mass tag)[6]标记的定量。这两种试剂都分为3个部分:反应基团、平衡基团和报告基团,如图1所示。反应基团在每组iTRAQ(以4标iTRAQ 为例)或TMT 试剂中的质量数是一致的,报告基团和平衡基团通过同位素的巧妙组合在每个标记试剂中各自质量数不相同但这两者的质量数之和相等,因此,这样3个部分的巧妙组合使得整个标记试剂成为“等重标记试剂”,即同一肽段经过含有不同同位素组合的试剂标记后,在一级质谱检测时质荷比完全相同,因此这样的标记不会增加一级谱图的复杂程度。具体而言,iTRAQ 或TMT 试剂中的反应基团用于将其标记在肽段上,只要有氨基端的肽段至少都会加上一个iTRAQ 或TMT 标签,另外赖氨酸(K)的侧链氨基也会被iTRAQ 或TMT 标签标记;在对一级质谱中带有标记的母离子(precursor ion)进行碎裂后,会产生低质荷比的报告离子,这些报告离子在二级质谱中的强度差异就代表了它所标记多肽的相对丰度;同时,二级质谱过程中多肽的酰胺键断裂,形成一系列b、y 离子,通过数据库查询和比较,可以鉴定出其相应的蛋白质。这类试剂中除了反应基团为NHS(N-羟基琥珀酰亚胺;该基团和肽段中的氨基反应)之外,还有反应基团是和半胱氨酸反应的等重同位素标记试剂如cysTMT[7]和iodoTMT[8],主要用于研究半胱氨酸上的修饰如S-亚硝基化修饰、氧化修饰和二硫键的连接等。这类等重同位素标记试剂用于蛋白质的定量有着明显的优点:第一,iTRAQ 试剂可以同时标记多达8个样品,TMT试剂则可以同时标记6个样品,因而一次实验即可实现多组样品之间的比较,提高了分析通量;第二,不同样品来源的同一蛋白质的同一个标记肽段在一级质谱上表现为一个峰,在不增加一级质谱复杂度的前提下,增强了一级质谱的信号,进一步提高了灵敏度;第三,有研究表明肽段被iTRAQ和TMT 标记后,二级图谱质量更好,产生更加丰富的b、y 离子系列使得蛋白质序列覆盖率提高,鉴定结果更可靠[9]。iTRAQ和TMT的普遍应用为大规模的蛋白质组定量提供了有利的工具,研究者们也对它们做了较为系统的评价。Pichler 等[10]以Hela 细胞为样本的研究表明:从蛋白质鉴定的角度而言,4标iTRAQ 比8标iTRAQ 得到近一倍以上的蛋白质鉴定结果,主要由于4标试剂和8标试剂在结构上有所差别,导致8标试剂在二级质谱中会产生母离子丢失标记试剂的不规则碎片,难以被常见的搜索引擎解析,对鉴定性能产生了负面影响;Ciborowski等[11]对用4标和8标iTRAQ 试剂分别标记的血浆样品定量分析,发现了8标的iTRAQ 试剂的定量结果偏差更小。Yates 等[12]对TMT 试剂的比较则表明,以6标TMT 中任意3标分别标记两组样品各进样一次和以2标TMT 标记后3次重复进样相比,两种方法获得的有定量信息的蛋白质数目相近,但使用6标TMT的样品中可获得更多的显著差异表达的蛋白质,并且缩短了分析时间。当然,基于等重标记结合报告离子的方法也有着一些缺陷,最大的缺陷是在产生信号的低质量范围有比较严重的信号抑制效应,影响了定量的动态范围和准确度,难以满足低丰度蛋白质的定量要求。此外,由于iTRAQ和TMT 试剂盒价格昂贵,反应步骤复杂也限制了它们的使用。
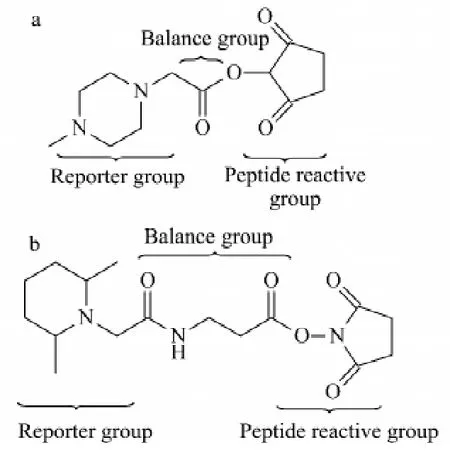
图1 (a)iTRAQ和(b)TMT 试剂的结构示意图Fig.1 Structures of(a)iTRAQ and(b)TMT
近年来,不断有更加廉价易得的等重同位素标记试剂被开发和应用。Li 等[13]发展了一种带有亲和标签的可断裂等重同位素标记试剂CILAT(cleavable isobaric labeled affinity tag)。该试剂由生物素亲和标签、酸可裂解键、报告基团、平衡基团以及巯基反应基团等部分共同组成。由于酪氨酸可以被酪氨酸酶氧化为邻醌,得到的邻醌可以和巯基通过迈克尔加成反应将CILAT 标记到肽段上,并且还可以利用所引入的亲和标签富集含酪氨酸的肽段;另外,酪氨酸在蛋白质中的含量(3.35%)远高于半胱氨酸(0.99%),因此这样的富集还可以提高蛋白质鉴定的序列覆盖度。之后,该课题组[14]又通过对同位素标记基团的改进,将该标记试剂由可对两组样品同时进行标记改进到可对12组样品同时进行标记。上述的标记试剂由于只和酪氨酸氧化后的邻醌反应,对肽段的定量有一定的偏向性。该课题组[15,16]最近又基于iTRAQ的反应基团设计了一种DiART(deuterium isobaric amine reactive tag)试剂。DiART试剂为6种同一结构的分子,包含报告基团、平衡基团和与iTRAQ和TMT 相同的反应基团NHS。每种DiART 试剂包含4个2H 原子,置于亲水基团附近,用于消除2H 原子造成的色谱流出差异效应。该试剂的合成步骤仅需6步,最终产率为30%~40%,大大优于TMT 试剂的14步合成路线及其低于1%的产率。同时在试剂中采用2H 原子代替价格昂贵的13C 或15N 原子也大大降低了成本。他们将DiART 试剂与电喷雾-线性离子阱-傅里叶变换静电场轨道阱(ESI-LTQ-Orbitrap)结合,采用碰撞诱导碎裂-高能碰撞碎裂(CID-HCD)方法对DiART 标记的肽段进行了高可信度的定性以及高准确度的定量,同时采用电子转移裂解-高能碰撞碎裂(ETD-HCD)方法对磷酸化肽段进行了定性定量分析。Li 等[17]发展了一种DiLeu(N,N-dimethyl leucine)试剂,该试剂包含能与肽段N 端和赖氨酸ε氨基反应的基团(三嗪酯)、平衡基团和报告基团。DiLeu 标记后的肽段比未标记的肽段相对分子质量增加145.1 Da,而在串级质谱中可分别产生m/z 115.1、116.1、117.1和118.1的报告离子。在用于蛋白质的酶解样品时,DiLeu和iTRAQ 试剂显示了类似的蛋白质序列覆盖度(~43%)以及定量准确性(<15%)。同时发现DiLeu 能够增强碎裂,为蛋白质鉴定和从头测序提高可信度。此外,DiLeu 合成所需要的试剂均为市售试剂,合成步骤简单,因此相比iTRAQ 试剂,费用大大降低。但该试剂的缺点是反应基团的普适性不够强,需要比较特殊的反应条件才能将标签标记于肽段上。Deshaies和Beauchamp 等[18]基于点击化学发展了一种以加州理工学院命名的等重同位素标记试剂CITs(caltech isobaric tags),通过铜(I)催化的叠氮-炔的环加成反应得到了气相可裂解的键用于产生报告离子,该试剂在二级质谱的碰撞碎裂过程中会释放出稳定的阳离子型报告基团,且将报告离子的质量数分别增加到164和169,可以降低三维离子肼在低质量端的cut-off 效应。
以上的方法中,一次实验可定量的样品数由定量试剂的报告离子数目决定,通常一次实验中可定量2~8组样品。对于更高通量的要求,有研究者将等重同位素标记和非等重同位素标记进行结合,在一级和二级质谱上同时进行定量,将一次定量的样品最多扩展到了12组[19]或18组[20];也有研究者将高精度质谱和等重同位素标记进行结合,利用高精度质谱对商业化试剂TMT 报告离子微小的质量差异(~6 mDa)进行进一步的区分,将6组标记的试剂扩展到实现8组样品的同时定量[21]。在蛋白质绝对定量的研究中,也有工作将二级质谱定量的方法和一级质谱定量的方法相结合,拓展了一次实验中可定量的样本数。我们课题组发展了“iTRAQ结合18O”标记相对定量技术[22],实现了4种不同样品中目标糖蛋白的糖基化位点占有率的同时定量。即首先采用4组iTRAQ 试剂对4组样品分别进行标记(标记试剂报告离子114~117),然后再任意选取其中2组用H216O 进行PNGaseF 去糖链处理(114,115标记的样品),另两组则在H218O 中进行去糖链处理(116,117标记的样品)。标记后进行混合,放大串联四极杆飞行时间质谱仪的母离子检测窗口,iTRAQ 标记试剂可在二级质谱中获得肽段的定量信息,16O 或18O 可在一级质谱中获得糖基化位点的定量信息,从而实现4组样品中糖基化位点程度的定量。
2.1.2 基于亚氨离子定量
肽段中缬氨酸、亮氨酸或异亮氨酸在二级质谱中容易形成亚氨离子(immonium ion),因此也可以用于发展成二级质谱定量方法。Quadroni 等[23]将细胞培养稳定同位素标记技术(SILAC)和等重标记技术相结合,通过在缬氨酸、亮氨酸或异亮氨酸的羰基和氨基中分别引入13C 或15N 标记(例如在这3种氨基酸残基中13C=O 对应的是14N,或12C=O 对应的是15N),由于这3个氨基酸都只增加1 Da 因此在细胞培养过程中可实现等重标记。在二级质谱中所形成的亚氨离子RC=NH 中N 原子由于存在轻、重同位素标记会产生1 Da的质量差异,这对亚氨离子的强度分别代表了两组样品中肽段的强度,从而可以实现二级质谱中的定量。
2.1.3 基于互补离子定量
基于二级质谱报告离子定量的技术在质谱提取母离子进行碎裂时,往往同时会引入其他色谱共流出相中相近质量数的干扰肽段,这些肽段在二级质谱中也会产生相同的报告离子,从而对目标肽段的定量准确性造成一定的影响。如果能使用带有肽段特征性信息的离子进行定量,则有助于提高定量的准确度。Gygi 等[24]最近报道了一种基于TMT 试剂标记的新型二级质谱定量方法,不同于常规的利用TMT的低质量端报告离子定量,他们使用的是TMTC(complement TMT fragment ion)离子用于定量。二级质谱用HCD 进行裂解时,TMT 标记的肽段碎裂产生报告离子后剩下的部分被称为TMTC离子,这样的TMTC离子与低质量端的报告离子具有等效的定量信息并带有肽段特征性信息。如图2所示,在一级质谱图中,干扰离子(蓝色)与目标离子(红色)同在分离窗口中,同时被选择裂解,若选择报告离子用于定量,由于干扰离子同样具有报告离子会造成定量的不准确,而使用互补的TMTC离子用于定量时由于TMTC离子的位置取决于目标肽段的质量与电荷数,去除了干扰离子的干扰,使得定量更加准确。他们将酵母酶解肽段以不同TMT 标记后按照一定比例混合作为目标肽段,TMT 标记的人源肽段作为干扰肽段,实现了蛋白质的准确定量。目前,TMTC最大的问题还是如何有效产生TMTC的标签用于定量,产生TMTC的效率取决于肽段的序列和电荷状态,还需要更深入的研究其规律性或者对TMT 试剂加以改进以保证产生尽可能多的TMTC离子。另外,由于TMT-129和TMT-130标记的肽段产生的TMTC质量数相同,在质谱中难以分辨,因此只能实现5组样品的定量。该课题组还开展了三级质谱定量的方法来消除色谱共洗脱质量数相近离子造成的定量误差[25]。样品经过Lys-C 酶解之后,生成的肽段在C-末端(K)和N-末端都带有氨基基团,与TMT 试剂反应会生成两端都带有TMT 标签的肽段。当母离子被提取进行二级质谱碎裂分析后,强度最强的碎片离子将进一步被提取进行三级质谱的碎裂。由于任意b、y 离子都含有TMT 标签,所以能够在三级质谱碎裂的过程中实现对目标肽段的相对定量。这一方法有效地消除了干扰肽段造成的定量误差,提高了定量的准确性。然而,基于三级质谱定量只是提高了TMT 所标记肽段本身的定量准确性,而一条肽段仍然只有一个定量信息,定量的随机误差不能避免。另外,由于需要在肽段两端都带上标记试剂,因此须采用Lys-C 酶切,这在一定程度上会导致蛋白质的鉴定率和定量率降低;额外的三级质谱数据采集也使得质谱分析流程更加复杂和耗时。
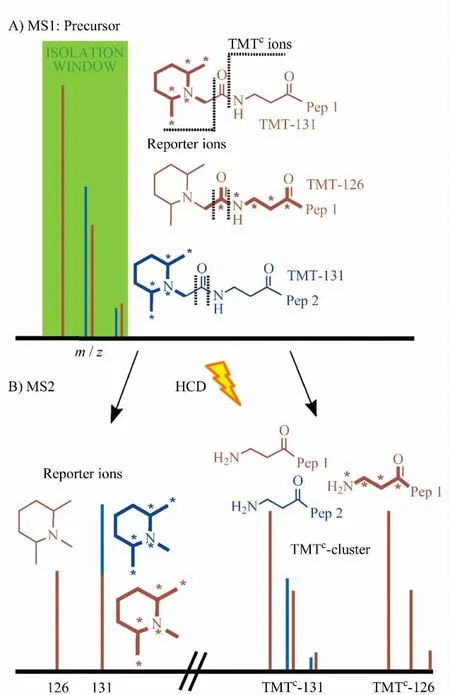
图2 互补离子(TMTC)定量方法的原理示意图Fig.2 Principle of interference-free quantification based on the TMTC cluster
2.2 基于多对离子定量
二级质谱过程中,产量最丰富的离子莫过于用于蛋白质鉴定的多对b、y 离子,如果这些离子对也带上质量标签,就可以被用于反映肽段量的变化。从单个离子代表一条肽段的定量信息发展为由多对离子反映一条肽段的定量信息,这样的改进提高了蛋白质的定量。多对离子定量方法的核心在于对肽段的C-末端和N-末端采用质量互补的同位素试剂标记产生等重标记的肽段,使得来源于不同样品的同一条肽段在一级质谱中质量数相同,在二级质谱中则产生成对的b、y 离子对用于肽段的定量和鉴定。这样的定量方法集合了开大窗口二级质谱定量和等重标记之报告离子二级质谱定量的优点,即通过等重标记无需开大窗口就可以实现不同标记肽段串级质谱共碎裂,且二级谱图中多对b、y 离子对用于肽段的定量有效降低了定量的随机误差,提高了定量的准确性。根据对肽段N-末端和C-末端引入标记的方法不同,目前的方法可以分为如下几类。
2.2.1 化学标记引入等重同位素标签
最早的等重标记b、y 离子对二级质谱定量是Thiede 等[26]发展的IPTL(isobaric peptide termini labeling)方法,是基于两步化学标记反应来实现肽段的等重标记。在Lys-C 酶解得到肽段后,分别在两组样品1和2中加入2-甲氧基-4,5-二氢-1H-咪唑(MDHI)-d4(重型)和MDHI(轻型)进行肽段C-末端标记反应,然后再加入琥珀酸酐(SA)(轻型)和SA-d4(重型)进行肽段N-末端标记反应,经过标记后不同样品中相同肽段实现了等重标记。该课题组[27]随后改变标记基团发展了另一种类似原理的标记方法,即Lys-C 酶解产生的肽段先进行N-末端SA(轻型)和SA-d4(重型)标记,然后分别进行肽段C-末端的重型和轻型二甲基化标记,使得标记流程更简单快捷,易于操作。Zou 等[28]则通过位点特异性反应分别在N-末端和C-末端引入标记,即首先在酸性条件下对N-末端进行二甲基化标记,然后将pH 调节至近中性,再实现C-末端赖氨酸上的二甲基化,两组样品即可通过调节二甲基化反应中的同位素组成实现等重标记。由于采用的是体外的化学标记,因此适用于多种样品来源(细胞、组织、体液等)的定量分析。但这类定量方法最大的缺陷在于,它们都采用了Lys-C 酶解以便产生以赖氨酸结尾的肽段用于C-末端的标记,与广泛使用的胰蛋白酶(trypsin)相比产生的肽段较长且带多电荷,Lys-C产生的肽段相对不利于基于碰撞诱导碎裂(CID)的质谱检测,导致肽段鉴定的效率降低;另外,含有赖氨酸漏切位点的肽段由于不能实现等重标记,不能用于b、y 离子对的二级质谱定量,导致了部分肽段定量信息的丢失。在最近的工作中,Thiede 等[29]将位点特异性反应和二甲基化同位素标记进行组合,在轻、重标记之间引入一组中等质量数标签,并且这组中等质量数标签使得肽段两端标记后的质量数增加和轻、重两组标记后的质量数增加一致,这样3组样品标记后在一级质谱上仍然只呈现一组峰,但在二级质谱中则呈现轻、中和重标记的成对b、y 离子,从而将等重标记b、y 离子定量的方法扩展到了3组。
2.2.2 代谢标记引入等重同位素标签
除了利用化学反应引入标记之外,选取合适的氨基酸加入到细胞培养基中进行细胞培养也可以引入等重标记,我们课题组[30]发展了一种新型的大规模的蛋白质标记定量方法,即体内终端氨基酸代谢标记(in vivo termini amino acid labeling,IVTAL)结合二级质谱定量的策略。如图3所示,该方法的核心是通过一组重同位素标记的精氨酸(13C6-R)和赖氨酸(13C6-K)分别进行细胞培养以及后续的特异性内切蛋白酶Lys-N和Arg-C的顺序酶解,得到以赖氨酸(K)开头和精氨酸(R)结尾的肽段,这些肽段在一级质谱中产生相同的母离子质量,但是在二级质谱的整个质量范围内可以产生具有6 Da 质量差异的b、y 碎片离子对,提取这些碎片离子对的峰强度可以用来定量相应的肽段和蛋白。同样由于是采用多对b、y 碎片离子对定量一条肽段,定量的随机误差被降低,因此IVTAL 方法保证了肽段定量的准确度。另外,由于IVTAL 方法中同位素的引入和样品的混合是在实验的早期,与SILAC 相似,实验误差被最小化。但这种方法主要适用于细胞来源样品的蛋白质的定量分析,并且需要使用Lys-N和Arg-C进行酶解。
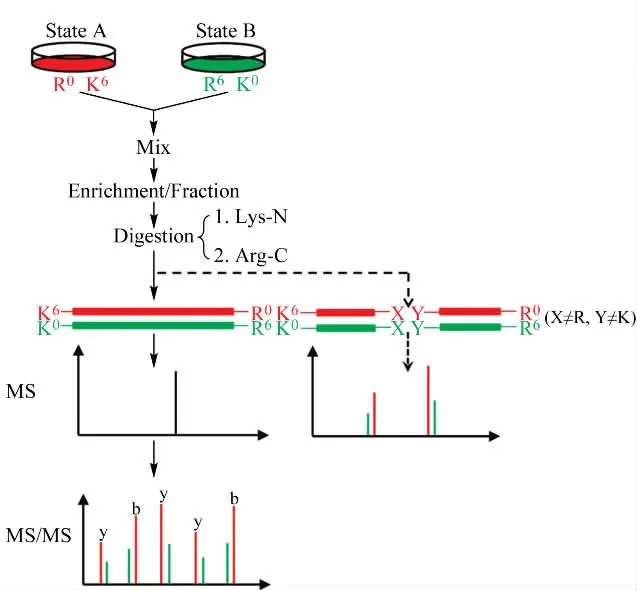
图3 体内终端氨基酸标记定量方法的流程图Fig.3 Schematic of in vivo termini amino acid labeling strategy(IVTAL)
2.2.3 化学标记和酶促标记组合引入等重同位素标签
我们课题组发展了一种化学标记和酶促标记结合的标记方法[31]。概括来说,两组对照样品的trypsin 酶解肽段分别在H216O 或H218O 水中进行trypsin催化的标记反应,所有以赖氨酸和精氨酸结尾的酶解肽段的C-末端都会被分别标记上两个16O 或18O原子,产生4 Da的差异。标记肽段经胍基化修饰后(目的是为了封闭侧链的氨基使其在下一步的二甲基化标记中不被标记),分别进行二甲基化标记,其中16O 标记的样品与氘代甲醛(CD2O)反应,18O 标记的样品与甲醛(CH2O)反应,这样两组样品中的相同肽段实现了等重标记,并且等重标记肽段进行反相色谱(RPLC)分离时同位素效应几乎可以忽略。该方法应用于鼠肝样品的定量蛋白质组分析结果表明,对于复杂样品,该方法具有很好的定量准确性。并且由于是胰酶进行的酶切,可定量的肽段比采用Lys-C 酶切的样品提高了近一倍,使得鉴定到的蛋白质中有84%的蛋白质有定量信息,而IVTAL和IPTL 方法仅有47%和42%鉴定的蛋白质有定量信息。
2.2.4 其他
针对目标蛋白质组的定量,Aebersold 等[32]建立了一种名为索引离子引发的二级质谱离子定量方法iMSTIQ(index-ion triggered MS2 ion quantification)。肽段用mTRAQ 试剂(Applied Biosystems)的+4Da的形式进行同位素标记,并合成了对应的在C 端R(+4 Da)和K(+8 Da)重标的目标样品肽段。合成的目标肽段中:一部分用mTRAQ的+0 Da的形式进行同位素标记作为参比肽段,该肽段与样品肽段mTRAQ的+4 Da 是等重的;另一部分用mTRAQ的+4 Da 或+8 Da的形式进行同位素标记作为“索引”肽段。目标肽段的样品肽段、参比肽段、索引肽段3种不同形式的化学性质相同,在LC 分析中可以共洗脱,在一级质谱分析中,样品和参比肽段均与索引肽段产生+8 Da的质量差异,通过索引肽段(加入时的含量较高)的检测引发目标样品、参比肽段的CID 过程,进而利用二级质谱中样品、参比肽段的多对b、y 离子相对强度的测定进行目标样品的定量分析。该方法的主要优势包括:1)通过含量较高的索引离子的检测引发目标肽段二级谱图的获得,不需要考虑目标肽段的离子强度。与基于inclusion list的目标蛋白质组学方法相比较,提高了灵敏度和准确性,对于复杂生物样品中低丰度目标肽段的定量分析具有独特的优势。2)与基于SRM的目标蛋白质组学方法相比较,无需预先选定最佳transition。3)利用二级质谱中多对特异性碎裂离子强度比进行定量,也增加了定量的准确性。但也由于索引肽段是针对目标肽段而合成的,所以这种方法不适合于大规模的蛋白质组定量。
3 多级质谱定量的发展趋势
大规模蛋白质组的相对定量需要更高准确度、更高精度、更高灵敏度和更高通量的新方法。多级质谱定量也正朝着这些目标努力。对于更高准确度的要求,有研究者将前体离子气相纯化技术和等重同位素标记相结合,通过质子转移离子-离子反应对前体离子先进行去卷积得到低电荷价态的相应离子再经过离子选择和碎裂,有效地排除了共洗脱相近质量数离子的干扰[33,34]。对于更高灵敏度的要求,则需要和多种蛋白质富集技术相结合,这就对富集技术的效率提出了更高的要求,要保证尽可能无偏向性、高回收率、高重复性地从复杂样品中富集到低丰度的蛋白质用于定量。对于翻译后修饰蛋白质组的定量而言,蛋白质水平的定量和修饰程度的定量都同样重要,等重标记的二级质谱b、y 离子定量的特点对于具有后修饰的蛋白质定量(尤其是富含多种后修饰类型和后修饰位点的肽段)可能会显现更大的优势,即产生的大量b、y 碎片离子可以对修饰位点进行具体的确认和对修饰程度进行定量,将克服一个肽段的强度包含了所有后修饰位点的变化情况的缺点。最后,对于这些新发展的蛋白质组多级质谱定量方法,还需要开发相应的生物信息学软件为所产生的质谱数据进行更加准确的解析。相信定量技术的不断发展会给大规模的差异蛋白质组研究提供更先进的研究策略,帮助科学家挖掘出更多重要的差异蛋白质。
[1]Ong S E,Mann M.Nat Chem Biol,2005,1(5):252
[2]Venable J D,Dong M Q,Wohlschlegel J,et al.Nat Methods,2004,1(1):39
[3]Zhang G,Neubert T A.Mol Cell Proteomics,2006,5(2):401
[4]White C A,Oey N,Emili A J.Proteome Res,2009,8(7):3653
[5]Ross P L,Huang Y N,Marchese J N,et al.Mol Cell Proteomics,2004,3(12):1154
[6]Thompson A,Schafer J,Kuhn K,et al.Anal Chem,2003,75(8):1895
[7]Murray C I,Uhrigshardt H,O'Meally R N,et al.Mol Cell Proteomics,2012,11(2):M111013441
[8]Iodoacetyl Tandem Mass TagTM(iodoTMTTM)Reagents.[2013-03-28].http://www.piercenet.com/instructions/2162387.pdf
[9]Gandhi T,Puri P,Fusetti F,et al.J Proteome Res,2012,11(8):4044
[10]Pichler P,Kocher T,Holzmann J,et al.Anal Chem,2010,82(15):6549
[11]Pottiez G,Wiederin J,Fox H S,et al.J Proteome Res,2012,11(7):3774
[12]Rauniyar N,Gao B,McClatchy D B,et al.J Proteome Res,2013,12(2):1031
[13]Li S,Zeng D.Chem Commun(Camb),2007,43(21):2181
[14]Zeng D,Li S.Bioorg Med Chem Lett,2009,19(7):2059
[15]Zeng D,Li S.Chem Commun(Camb)2009,45(23):3369
[16]Zhang J,Wang Y,Li S.Anal Chem,2010,82(18):7588
[17]Xiang F,Ye H,Chen R,et al.Anal Chem,2010,82(7):2817
[18]Sohn C H,Lee J E,Sweredoski M J,et al.J Am Chem Soc,2012,134(5):2672
[19]Robinson R A,Evans A R.Anal Chem,2012,84(11):4677
[20]Dephoure N,Gygi S P.Sci Signal,2012,5(217):rs2
[21]Werner T,Becher I,Sweetman G,et al.Anal Chem,2012,84(16):7188
[22]Zhang S,Liu X,Kang X,et al.Talanta,2012,91(14):122
[23]Colzani M,Schutz F,Potts A,et al.Mol Cell Proteomics,2008,7(5):927
[24]Wuhr M,Haas W,McAlister G C,et al.Anal Chem,2012,84(21):9214
[25]Ting L,Rad R,Gygi S P,et al.Nat Methods,2011,8(11):937
[26]Koehler C J,Strozynski M,Kozielski F,et al.J Proteome Res,2009,8(9):4333
[27]Koehler C J,Arntzen M O,Strozynski M,et al.Anal Chem,2011,83(12):4775
[28]Qin H,Wang F,Zhang Y,et al.Chem Commun(Camb),2012,48(50):6265
[29]Koehler C J,Arntzen M O,de Souza G A,et al.Anal Chem,2013,85(4):2478
[30]Nie A Y,Zhang L,Yan G Q,et al.Anal Chem,2011,83(15):6026
[31]Yang S J,Nie A Y,Zhang L,et al.Journal of Proteomics,2012,75(18):5797
[32]Yan W,Luo J,Robinson M,et al.Mol Cell Proteomics,2011,10(3):M110.005611
[33]Wenger C D,Lee M V,Hebert A S,et al.Nat Methods,2011,8(11):933
[34]Dayon L,Sonderegger B,Kussmann M.J Proteome Res,2012,11(10):5081
