基于稳定同位素标记的蛋白质组学定量方法研究进展
2013-07-13周愿单亦初张丽华张玉奎
周愿,单亦初,张丽华*,张玉奎
(1.中国科学院大连化学物理研究所,中国科学院分离分析化学重点实验室,国家色谱研究分析中心,辽宁大连 116023;2.中国科学院大学,北京 100039)
对细胞、组织和整个生物体的表达蛋白质进行定量分析是后基因时代面临的主要挑战之一[1]。蛋白质组定量分析可以分为相对定量和绝对定量[2]。相对定量是研究不同情况下同一蛋白质组样品组成含量的变化,如细胞受到刺激前后蛋白质表达的差异;绝对定量是研究样品中目标蛋白质的具体量,如拷贝数或浓度。
早期的蛋白质组定量主要利用双向凝胶电泳或双向差异凝胶电泳来实现。虽然该方法对蛋白质分离具有较高的分辨率,然而无法对疏水、低丰度和翻译后修饰蛋白质进行定量。近年来基于液相色谱-串级质谱联用的方法得到了快速发展,已成为蛋白质组定量的主流技术。该方法可以分为无标记定量法和稳定同位素标记定量法。其中,基于稳定同位素标记的蛋白质组定量方法可以在不同步骤实现样品混合,可以同时实现多重标记及消除色谱-质谱联用分析过程中不稳定性带来的定量误差等优点,已成为定量蛋白质组学研究中最常用的方法。
根据同位素引入的方式,基于稳定同位素标记的蛋白质组定量方法可以分为代谢标记法、化学标记法和酶解标记法。采用不同方法,标记同位素的样品在不同步骤混合;越早混合,样品预处理步骤引入的误差越小,定量的准确度越高。进行相对定量时,采用代谢标记法则样品在细胞水平混合;采用化学标记法则样品在蛋白质或者肽段水平混合;采用酶解标记法则样品在肽段水平混合。进行绝对定量时,样品在蛋白质或者肽段水平混合(见图1)。本文结合近期发表的文献,对基于稳定同位素标记的蛋白质组定量方法进行综述和评述。
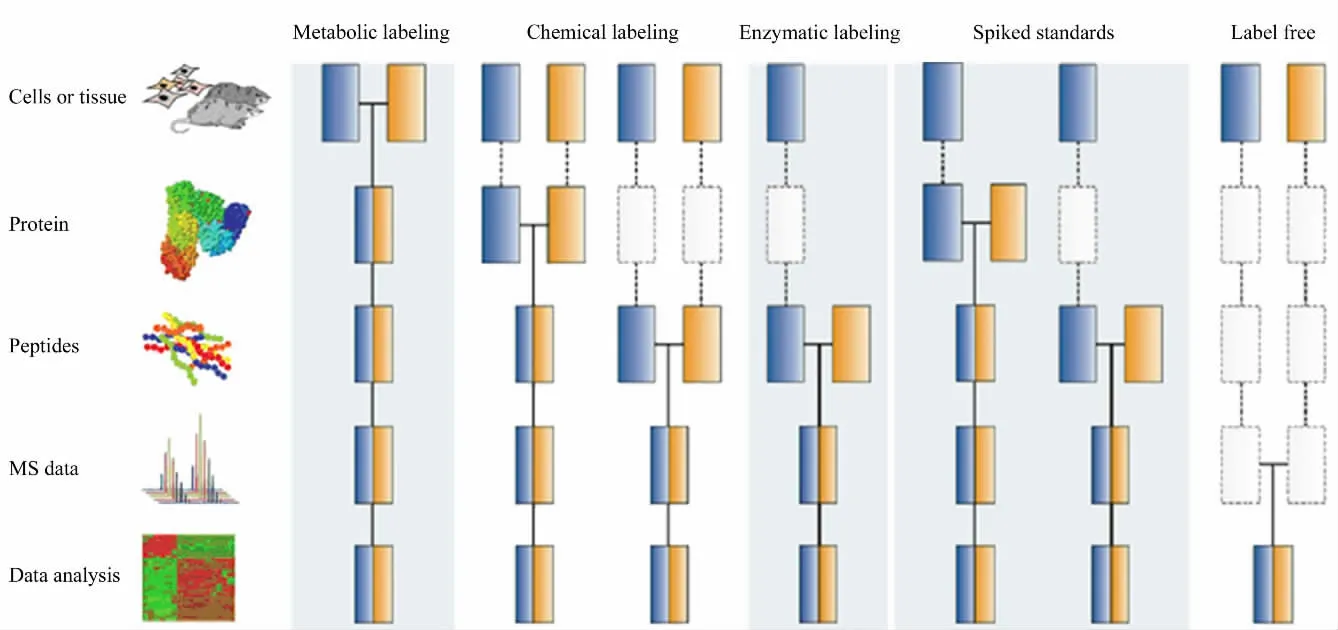
图1 基于质谱的蛋白质组学定量方法及实验流程(改编自文献[2,3])Fig.1 Mass spectrometry-based quantitative proteomics methods and workflows(adapted from references[2,3])
1 基于稳定同位素标记的相对定量方法
基于稳定同位素标记的蛋白质组相对定量方法分为基于一级质谱(如细胞培养稳定同位素标记(SILAC)[4]、二甲基化标记[5]等)和串级质谱(如等重同位素标记相对与绝对定量方法(iTRAQ)[6]、等重肽末端标记方法(IPTL)[7]等)的定量方法。前者通过比较轻重标记的样品在一级质谱的峰强度/峰面积实现蛋白质组的相对定量分析;后者通过比较样品在二级或者三级质谱的特征性碎片离子的峰强度实现蛋白质组的相对定量分析。
1.1 基于一级质谱的相对定量方法
适用于基于一级质谱的相对定量方法的稳定同位素标记试剂种类多、数据分析软件对其的支持较好,因此目前得到了较为广泛的使用。常用的方法有SILAC、15N 标记[8]、二甲基化标记、质量差异同位素标记相对与绝对定量标签(mTRAQ)标记[9]、18O标记[10]等。其中SILAC和15N 为代谢标记;二甲基化标记和mTRAQ 为化学标记;18O 标记为酶解标记。
1.1.1 基于代谢标记的相对定量方法
代谢标记是指在细胞或生物体成长过程加入含有稳定同位素标记的培养基,完成细胞或生物体标记的方法。常见的代谢标记方法有SILAC和15N 标记法,尤其SILAC 法使用更为广泛。该方法是在细胞培养过程中加入稳定同位素标记的必需氨基酸,如赖氨酸(K)和精氨酸(R),使得每条肽段相差的质量数恒定。与15N 方法相比,由于肽段的质量差异数与氨基酸种类和数目无关,因此简化了相对定量分析的难度。
为了解决SILAC 法只适用于细胞样品分析的不足,发展了培养基衍生同位素标签(CDITs)[11]、同位素标记蛋白质组(SILAP)[12]和超级SILAC(super-SILAC)[13]等方法。这些方法采用稳定同位素标记的细胞样品作为内标,通过与不同状态的组织样品混合,实现了组织样品蛋白质组表达的差异分析。此外,为了便于考察在体内蛋白质组的动态变化,如蛋白质的生成和降解,脉冲SILAC(pulsed SILAC)[14,15](见图2)应运而生。该方法首先将经过不同刺激的细胞在轻标记的培养液中培养,之后分别转入中标记和重标记的培养液中培养;通过比较中标记和重标记的比值,实现不同刺激条件下蛋白质表达量的差异分析。或将在轻标记培养液中培养的细胞分成两份,分别接受不同的刺激,并分别进行中标记和重标记;再通过比较中、重标记的比值,实现不同刺激条件下蛋白质表达量的差异分析[15]。
为降低同位素峰的干扰对定量的影响,基于一级质谱的定量方法通常要求MS1的质量差大于4 Da。近期,Hebert 等[16]提出了中子编码SILAC 法(NeuCode SILAC);利用13C、15N和2H 同位素因核结合能而导致的质量亏损,通过高分辨(分辨率(Rs)>200000)质谱对这细微的差异进行分析,实现了采用不同稳定同位素标记的两种蛋白质组样品的相对定量分析。该方法理论上可以实现多重样品的标记,但是对质谱的分辨率要求极高。

图2 脉冲SILAC 原理示意图[14,15]Fig.2 Principle of pulsed SILAC[14,15]
虽然基于代谢标记的方法在定量蛋白质组领域得到了极为广泛的使用,但是仍有一些不足:1)成本较高;2)难以用于人的组织样品及体液的分析;3)同位素标记氨基酸的使用会导致标记蛋白质的表达不同于非标记蛋白质的表达[17];4)在细胞培养过程中存在精氨酸向脯氨酸转化的现象。
1.1.2 基于化学标记的相对定量方法
除代谢水平标记外,通过体外化学标记引入同位素是一种非常有价值的蛋白质组相对定量方法;适用于细胞、体液、组织等多种样品分析。现有的化学标记试剂多数通过与氨基或巯基反应引入稳定同位素。最常用的是基于N-羟基琥珀酰胺(NHS)化学和还原胺反应,如mTRAQ和二甲基化标记,其中二甲基化标记使用更为广泛。
Hsu 等[5]发展的二甲基化标记是利用甲醛和氰基硼氢化钠组合标记肽段所有的活性氨基。该方法具有反应条件温和、快速、高效、无明显副反应、同位素试剂种类多、价格低廉等优点。Raijmakers 等[18]使用预柱进行在线标记,不仅减少了标记时间,而且降低了人为因素带来的误差,提高了定量的准确性和精密度。该课题组[19]还实现了在线二甲基化标记与二维液相色谱-基质辅助激光解吸-飞行时间质谱(2DLC-MALDI-TOF/TOF MS)的联用。此外,Wang 等[20]在固相标记后利用RPLC-SCX-RPLCESI-MS/MS 平台分析了人肝癌样品与正常组织样品,发现了94种蛋白质在肝癌样品中上调,249种蛋白质在肝癌样品中下调。Boersema 等[21]建立了二甲基化标记的标准流程,并分别建立了固相萃取柱标记、溶液标记和在线反相预柱标记三种方法。Qin 等[22]以磷酸化功能修饰的有序介孔有机硅为吸附剂,实现了内源性磷酸化肽段的富集和原位二甲基化标记,并成功用于肝癌病人和正常人血液中内源性磷酸肽的相对定量分析。Sun 等[23]在利用酰肼球富集糖肽的同时,进行糖肽的原位二甲基化标记,实现了高准确、高回收率的糖蛋白质组相对定量分析。
利用二甲基化标记试剂的组合可以实现样品的多重标记,进而实现多种样品的蛋白质组相对定量。Boersema 等[24]利用CH2O和NaCNBH3、CD2O和NaCNBH3以及13CD2O和NaCNBD33种标记试剂的组合,实现了蛋白质胰蛋白酶酶解产物的三重标记。Song 等[25]用轻、重试剂标记相同的样品,中标试剂标记另一份样品,实现了准三重标记;通过比较轻重标记,一次运行可得到两个定量结果,实现了高准确度和高精度的蛋白质组相对定量分析。Hsu 等[26]利用CH2O、CD2O、NaCNBH3和NaCNBD34种试剂组合标记Lys-C 酶解的肽段,实现了4种样品的同时分析;利用CH2O、CD2O、13CD2O、NaCNBH3和NaCNBD3有机组合标记Lys-C 酶解产物,还可以实现五重样品的同时分析。
1.1.3 基于酶解标记的相对定量方法
18O 标记是目前酶解标记的唯一方法[10]。采用该方法仅需要在酶解过程中使用H218O。使用的蛋白酶可以为胰蛋白酶(trypsin)、胰凝乳蛋白酶(chymotrypsin)、Glu 蛋白酶(Glu-C)和多肽内切酶(Lys-C)等丝氨酸蛋白酶类;在这些酶的催化下,可以在肽段C 端增加2个18O 原子。18O 标记既可用于非修饰蛋白质组的相对定量,而且也可以将肽段末端的18O 标记与去糖链过程的18O 标记相结合,实现糖肽位点和糖链的相对定量分析[27,28]。此外,将iTRAQ和18O 标记结合,还可以在实现糖蛋白质样品多重定量的同时,实现糖肽位点的确认[29]。将18O与二甲基化标记相结合,还可基于等重标记实现蛋白质组的高精度和高准确度的相对定量分析[30]。
近期基于固定化酶反应器的酶解也逐渐被用于酶解标记。该方法不仅能减少标记过程中回交的发生,还能显著提高标记效率,缩短标记时间[31,32]。
1.2 基于串级质谱的相对定量方法
尽管基于一级质谱的蛋白质组定量方法种类多,应用广泛,但是仍存在以下问题:1)定量的准确度受噪音离子的干扰,造成低峰强度组分的定量结果不准;2)定量动态范围低;3)难以实现同时超过四重标记。为解决上述问题,近年来基于串级质谱的蛋白质组相对定量方法受到了越来越多的关注。
基于串级质谱的蛋白质组相对定量方法可以分为基于报告离子和碎片离子的两种定量方法。前者主要是使用等重标记试剂,如iTRAQ和串联质量标签(TMT)[33]等,通过二级谱或三级谱的报告离子强度实现定量;后者则利用不同标记肽段的成对的碎片离子的强度实现定量。
1.2.1 基于报告离子的相对定量方法
iTRAQ[6]和TMT[33]是最常用的基于报告离子的蛋白质组相对定量方法。其中利用iTRAQ 试剂可以实现八重标记,利用TMT 试剂可以实现六重标记。通过方法改进或者与其他方法结合,可实现更多重的标记,进而提高蛋白质组相对定量的通量,是该类方法的研究目标之一。Dephoure 等[34]将三重SILAC 标记结合六重TMT 标记,实现了18种样品的同时分析。Werner 等[35]和McAlister 等[36]利用高分辨质谱将TMT 试剂由六重增加为八重,显著提高了该方法的分析通量。近期也发展了一些新的基于报告离子的相对定量方法,包括N,N-二甲基化亮氨酸定量标签(N,N-dimethyl leucines,DiLeu)[37]、氘代等重氨基反应标签(deuterium isobaric amine-reactive tags,DiART)[38]、加州理工等重标记(Caltech isobaric tags,CIT)[39]、可裂解等重亲和标签(cleavable isobaric labeled affinity tag,CILAT)[40,41]。其中CIT 可以根据需要设计试剂,实现n 重标记;CILAT单次实验可以实现十二重标记。
然而,等重标记方法的不足在于,共洗脱组分和目标组分在碎裂池内共碎裂,会影响定量的准确度,存在所谓的低估效应(underestimation),即在复杂生物样品分析中,共碎裂的离子产生的报告离子会干扰目标母离子在二级谱中报告离子的强度,使得实验中得到蛋白质的定量结果低于真实值。为了解决该问题,Gygi 等[42]将二级谱中高强度的碎片离子进一步碎裂,利用三级谱中的报告离子强度进行定量。除此以外,Gygi 等[43]利用TMT 标记在二级谱中会产生与TMT 报告离子互补的离子——TMT 互补离子(TMTC),由于TMTC特异性强,通过去卷积处理也可以实现肽段的准确定量。
1.2.2 基于碎片离子的相对定量方法
基于碎片离子的相对定量方法也可以解决基于报告离子的相对定量方法存在的低估效应。该方法将肽段的N 端和C 端分别用轻标和重标试剂标记,使肽段在一级质谱上具有相同或相似的m/z 而无法分辨,在二级谱碎裂时会形成成对的碎片离子,利用这些碎片离子强度之比可以实现蛋白质的相对定量分析。等重肽末端标记方法(IPTL)[7,44,45]是该类方法的第1个实例,并已经实现了3次技术更新,分别利用SA(succinic anhydride)及其同位素形式标记肽段的N 端氨基、MDHI(2-methoxy-4,5-dihydro-1Himidazole)及其同位素形式标记肽段的C 端赖氨酸上的氨基、利用2种肽段鉴定时得分值的不同实现肽段和蛋白质的相对定量[7]。为了提高标记效率和速度,该课题组[45]又开发了第二代IPTL。使用SA 标记肽段的N 端氨基,同时使用甲醛标记肽段的C 端赖氨酸上的氨基,并开发了基于强制搜索的IsobariQ 定量软件。最近,为进一步提高相对定量的通量和选择性,该课题组[44]利用二甲基化标记方法在酸性条件标记肽段的N 端,碱性条件下标记肽段的C 端赖氨酸上的氨基,并利用甲醛标记试剂种类多的特点实现了三重标记(见图3)。这也是该类方法唯一实现多重标记的实例。
等重末端标记定量方法(QITL)[30]利用甲醛标记肽段的N 端氨基,18O 标记肽段的C 端(当C 末端为K 时需要胍基化),从而实现等重标记。该方法定量覆盖率接近85%,为现有该类方法中定量覆盖率最高的方法。
除化学标记外,代谢水平标记也可以实现肽段的等重标记。体内终端氨基酸代谢标记(IVTAL)[46]是在代谢水平实现等重标记的实例之一。该方法在细胞培养时用(K6,R0)和(K0,R6)分别培养不同的细胞;混合后用Lys-N和Arg-C 两种酶对蛋白质进行酶切,其中一部分肽段会产生N端为K、C 端为R的序列。这些肽段在一级质谱上没有差异,而碎裂时会产生明显的碎片离子对,进而实现蛋白质组的相对定量。索引离子触发二级谱离子定量方法(iMSTIQ)[47]是利用代谢标记和化学标记相结合的方法实现等重标记。
基于碎片离子的蛋白质组相对定量方法的优点在于,可以克服使用离子阱类质谱的1/3效应,通过使用特征性的碎片离子,提高定量的准确度和精密度。然而该方法同样存在标记步骤繁多、标记选择性和反应效率有待提高,以及二级质谱复杂度成倍增加会降低肽段得分、进而减少可定性和定量的蛋白质数目等问题。
2 基于稳定同位素标记的绝对定量方法

图3 Triplex-IPTL 流程图[44]Fig.3 Triplex-IPTL approach[44]
基于上述方法,实现上万蛋白质的相对定量分析已成为现实。然而,对于临床诊断等,仅仅知道有哪些蛋白质是差异蛋白质尚无法满足要求,仍需要知道目标蛋白质的绝对定量信息。
基于抗体的绝对定量方法(如酶联免疫吸附测定(ELISA))具有很高的选择性和灵敏度,但是受限于抗体种类,且分析通量有待提高。基于质谱的绝对定量方法能够实现多种目标蛋白质的定量,因此受到了广泛的关注。其中,基于稳定同位素标记的绝对定量方法是向待测样品中加入已知量的同位素标记的内标肽段、蛋白质或由多种目标肽段构建的串联体,并以此衡量样品中目标蛋白质的量。
基于稳定同位素标记肽段为内标的绝对定量方法包括绝对定量法(AQUA)[48]、18O 标记[31]、二甲基化标记肽段[49]或者mTRAQ 标记肽段[9];同位素标记蛋白质为内标的绝对定量方法包括全序列重标记蛋白质标准物绝对定量法(PSAQ)[50]、绝对SILAC 方法(absolute SILAC)[51]和全序列表达稳定同位素标记蛋白(FlexiQuant)[52]等;同位素标记的串联体为内标的绝对定量方法包括定量串联体(Qcon-CAT)[53]、等电串联体(iQCAT)[54]等;基于目标蛋白质片段的绝对定量方法包括蛋白抗原决定基标签(PrEST)[55]等。上述方法的实验流程[56]如图4所示。由于内标与样品混合的步骤越靠前,绝对定量的准确度越高,因此理想的内标应该是同位素标记的蛋白质;该蛋白质应具有与目标蛋白质相同的三级和四级结构、相同的翻译后修饰和已知的蛋白质浓度[57]。
2.1 以同位素标记肽段为内标物的绝对定量方法
AQUA 方法[48]是将待测样品酶解后加入已知量的人工合成的含有稳定同位素标记的肽段内标物或者用化学法标记上同位素的肽段内标物,通过比较目标肽段和添加内标物的信号强度,得到目标肽段的含量。由于该方法内标肽容易从商业公司获取,因而在绝对定量中得到了极为广泛的应用。基于相同的原理,在体外对目标肽段进行同位素标记,如18O[31]、二甲基化[49]或者mTRAQ[9]标记的肽段,也可以作为内标物加入到待测体系,用于实现目标蛋白质的定量。
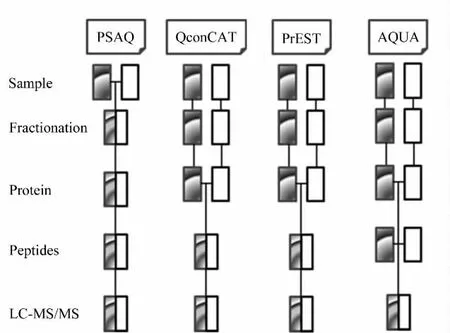
图4 基于稳定同位素稀释的蛋白质绝对定量分析流程图(改编自文献[56])Fig.4 Stable isotope dilution strategies for absolute quantification of targeted proteins(adapted from reference[56])
然而由于该方法是在待测样品酶解后加入内标物,忽视了样品前处理过程引入的损失,因此定量结果有不确定性。此外,该方法成本较高,难以实现大量目标蛋白质的绝对定量分析。
2.2 以同位素标记蛋白质为内标物的绝对定量方法
除了以标记肽段为内标外,还可以将目标蛋白质全序列进行同位素标记,如PSAQ[50],absolute SILAC[51]和FlexiQuant[52]等方法。通过这些方法,可以在细胞外或者细菌体内代谢合成全长的同位素标记蛋白质。纯化后的蛋白质可以直接加入到细胞提取液中,实现待测样品中目标蛋白质的绝对定量。尽管采用该方法可以消除所有样品处理过程引入的定量不确定性,但是仅适用于可溶性蛋白质的定量分析。此外,标记过程较为复杂,难以实现大规模目标蛋白质的绝对定量。
2.3 以同位素标记串联体为内标物的绝对定量方法
QconCAT[53]利用了重组DNA 技术,将多种目标蛋白质的目标肽段模拟构建成新的蛋白质,并将对应的质粒转入到细菌中,在含有同位素的培养液中培养,从而引入同位素标记蛋白质。这些蛋白质经纯化后,在酶解前加入到待测样品的蛋白质提取液中。该方法不仅可以同时实现多种蛋白质的绝对定量,而且可以消除酶解过程引入的误差。尽管在理论上可以有效地产生目标肽段,但是该蛋白质与目标蛋白质的结构不同,酶解效率也可能存在差别。此外,由于该方法无法用于蛋白质样品酶解前的分级过程,通常一种蛋白质只能用一条肽段去定量,缺乏统计意义。
2.4 基于目标蛋白质片段的绝对定量方法
Mann 等[55]结合人PrEST与SILAC 方法,实现了目标蛋白质的绝对定量。首先将PrEST 在E.coli体内表达、纯化并定量后,加入到SILAC 标记的样品中,并通过比较两者的强度实现目标的定量。该方法不仅可以实现多种蛋白质的同时定量,而且由于内标物可以在样品预处理过程中加入,提高了定量的准确度。
3 结论与展望
现有的蛋白质组定量方法种类繁多,使用范围广泛,加速了生命科学的研究进程。然而采用这些方法时,大部分实验过程都是离线操作。如何减少人为操作,实现样品定量分析的在线化、集成化,进而提高定量的通量、准确度和精密度是该领域的发展方向之一。尽管已有许多商品化标记试剂,但是仍需要不断发展新型标记试剂,并使其具有标记方法简单、高效、无副反应、标记后处理简单、定量结果准确、精密度高、动态范围宽、适用的样品类型范围广等优点。此外,提高LC-MS,尤其是nanoLC-ESI MS的重现性,对于提高定量结果的精密度至关重要。质谱仪扫描速度、质量精度和分辨率的不断提高,也必将为蛋白质组定量新方法的发展提供新的机遇。
目前稳定同位素标记的蛋白质组定量方法尚存在一些挑战,包括如何实现低丰度蛋白质和蛋白质异构体的定量分析、未知基因库的生物蛋白质表达谱和翻译后修饰的动态变化分析等。这些问题的解决,都亟须蛋白质组定量新试剂、新技术和新方法的不断发展。
[1]Bantscheff M,Lemeer S,Savitski M M,et al.Anal Bioanal Chem,2012,404(4):939
[2]Ong S E,Mann M.Nat Chem Biol,2005,1(5):252
[3]Bantscheff M,Schirle M,Sweetman G,et al.Anal Bioanal Chem,2007,389(4):1017
[4]Ong S E,Blagoev B,Kratchmarova I,et al.Mol Cell Proteomics,2002,1(5):376
[5]Hsu J L,Huang S Y,Chow N H,et al.Anal Chem,2003,75(24):6843
[6]Ross P L,Huang Y N,Marchese J N,et al.Mol Cell Proteomics,2004,3(12):1154
[7]Koehler C J,Strozynski M,Kozielski F,et al.J Proteome Res,2009,8(9):4333
[8]Oda Y,Huang K,Cross F R,et al.Proc Natl Acad Sci U S A,1999,96(12):6591
[9]DeSouza L V,Taylor A M,Li W,et al.J Proteome Res,2008,7(8):3525
[10]Yao X,Freas A,Ramirez J,et al.Anal Chem,2001,73(13):2836
[11]Ishihama Y,Sato T,Tabata T,et al.Nat Biotechnol,2005,23(5):617
[12]Yu K H,Barry C G,Austin D,et al.J Proteome Res,2009,8(3):1565
[13]Geiger T,Cox J,Ostasiewicz P,et al.Nat Methods,2010,7(5):383
[14]Selbach M,Schwanhausser B,Thierfelder N,et al.Nature,2008,455(7209):58
[15]Schwanhausser B,Gossen M,Dittmar G,et al.Proteomics,2009,9(1):205
[16]Hebert A S,Merrill A E,Bailey D J,et al.Nat Methods,2013,10(4):332
[17]Filiou M D,Varadarajulu J,Teplytska L,et al.Proteomics,2012,12(21):3121
[18]Raijmakers R,Berkers C R,de Jong A,et al.Mol Cell Proteomics,2008,7(9):1755
[19]Raijmakers R,Heck A J,Mohammed S.Mol BioSyst,2009,5(9):992
[20]Wang F,Chen R,Zhu J,et al.Anal Chem,2010,82(7):3007
[21]Boersema P J,Raijmakers R,Lemeer S,et al.Nat Protoc,2009,4(4):484
[22]Qin H,Wang F,Wang P,et al.Chem Commun(Camb),2012,48(7):961
[23]Sun Z,Qin H,Wang F,et al.Anal Chem,2012,84(20):8452
[24]Boersema P J,Aye T T,van Veen T A,et al.Proteomics,2008,8(22):4624
[25]Song C,Wang F,Ye M,et al.Anal Chem,2011,83(20):7755
[26]Hsu J L,Huang S Y,Chen S H.Electrophoresis,2006,27(18):3652
[27]Shakey Q,Bates B,Wu J.Anal Chem,2010,82(18):7722
[28]Liu Z,Cao J,He Y,et al.J Proteome Res,2009,9:227
[29]Zhang S,Liu X,Kang X,et al.Talanta,2012,91:122
[30]Yang S J,Nie A Y,Zhang L,et al.J Proteomics,2012,75(18):5797
[31]Qin W,Song Z,Fan C,et al.Anal Chem,2012,84(7):3138
[32]Mirza S P,Greene A S,Olivier M.J Proteome Res,2008,7(7):3042
[33]Thompson A,Schafer J,Kuhn K,et al.Anal Chem,2003,75(8):1895
[34]Dephoure N,Gygi S P.Sci Signal,2012,5(217):rs2
[35]Werner T,Becher I,Sweetman G,et al.Anal Chem,2012,84(16):7188
[36]McAlister G C,Huttlin E L,Haas W,et al.Anal Chem,2012,84(17):7469
[37]Xiang F,Ye H,Chen R,et al.Anal Chem,2010,82(7):2817
[38]Zhang J,Wang Y,Li S.Anal Chem,2010,82(18):7588
[39]Sohn C H,Lee J E,Sweredoski M J,et al.J Am Chem Soc,2012,134(5):2672
[40]Li S,Zeng D.Chem Commun,2007(21):2181
[41]Zeng D,Li S.Bioorg Med Chem Lett,2009,19(7):2059
[42]Ting L,Rad R,Gygi S P,et al.Nat Methods,2011,8(11):937
[43]Wühr M,Haas W,McAlister G C,et al.Anal Chem,2012,84(21):9214
[44]Koehler C J,Arntzen M O,de Souza G A,et al.Anal Chem,2013,85(4):2478
[45]Koehler C J,Arntzen M O,Strozynski M,et al.Anal Chem,2011,83(12):4775
[46]Nie A Y,Zhang L,Yan G Q,et al.Anal Chem,2011,83(15):6026
[47]Yan W,Luo J,Robinson M,et al.Mol Cell Proteomics,2011,10(3):M110.005611
[48]Gerber S A,Rush J,Stemman O,et al.Proc Natl Acad Sci U S A,2003,100(12):6940
[49]Ji C,Sadagopan N,Zhang Y,et al.Anal Chem,2009,81(22):9321
[50]Brun V,Dupuis A,Adrait A,et al.Mol Cell Proteomics,2007,6(12):2139
[51]Hanke S,Besir H,Oesterhelt D,et al.J Proteome Res,2008,7(3):1118
[52]Singh S,Springer M,Steen J,et al.J Proteome Res,2009,8(5):2201
[53]Rivers J,Simpson D M,Robertson D H,et al.Mol Cell Proteomics,2007,6(8):1416
[54]Austin R J,Chang D K,Holstein C A,et al.Proteomics,2012,12(13):2078
[55]Zeiler M,Straube W L,Lundberg E,et al.Mol Cell Proteomics,2012,11(3):O111.009613
[56]Rodríguez-Suárez E,Whetton A D.Mass Spectrom Rev,2013,32(1):1
[57]Simpson D M,Beynon R J.Anal Bioanal Chem,2012,404(4):977
