应用溶蚀器/后置膜系统分析上海大气PM2.5中水溶性离子的组成及采样误差
2013-06-26管晶晶龚识懿冯加良
张 攀, 仲 勉, 管晶晶, 荆 亮,李 曼,龚识懿,冯加良*
应用溶蚀器/后置膜系统分析上海大气PM2.5中水溶性离子的组成及采样误差
张 攀, 仲 勉, 管晶晶, 荆 亮,李 曼,龚识懿,冯加良*
(上海大学 环境与化学工程学院 环境污染与健康研究所, 上海 200444)
PM2.5; 水溶性离子; 采样误差; 氨; 上海
0 引 言
PM2.5(空气动力学直径小于2.5 μm的大气颗粒物, 也称为大气细颗粒物)是我国当今面对的主要的大气污染物, 对人体健康有很大的危害[1], 正受到越来越多的关注。细颗粒物也是导致大气灰霾频发的主要因素, 其中水溶性离子是散射可见光的主要物质[2]。同时, PM2.5的干湿沉降已被认识到是水体/湖泊营养物质的重要来源[3]。因此, 了解PM2.5的化学组成是准确评价大气颗粒物环境影响的基础。虽然基于颗粒物吸湿增长特性的实时在线方法近年来得到了快速发展, 并已被用于PM2.5化学组成的分析[4], 但已有研究发现在某些情形下在线方法会产生较大的误差[5]。膜过滤方法仍被绝大多数的研究所采用, 且采样时间通常超过12 h。采样过程中颗粒-颗粒之间、气体-颗粒之间的相互作用以及半挥发性组分的分解等会导致采样误差的发生, 如铵根离子、硝酸根离子、氯离子和硫酸根离子等主要水溶性离子均可能存在采样误差[6]。但对膜过滤方法的采样误差的大小及影响因素仍有很多不明确的地方, 尤其是针对国内环境条件的采样误差分析。本研究用溶蚀器(denuder)/后置膜(backup-filter)采样系统对上海市区的PM2.5及气态组分进行采集, 拟分析PM2.5中主要水溶性离子的浓度与分布, 探讨常规采样过程中的采样误差及其影响因素。
1 材料与方法
1.1 样品采集
采样地点为上海大学闸北校区科技楼楼顶, 距离地面约30 m, 采集时间为2008年的9月、10月和2009年的1月、9月, 每次采集6 d的样品, 每个样品的采集时间为24 h。4次采样期间的大气日平均温度分布范围分别为24~27 ℃、16~24 ℃、-1~7 ℃和22~27 ℃, 平均26 ℃、19 ℃、4 ℃和25 ℃; 相对湿度日均值范围分别为68%~82%、58%~81%、51%~87%和66%~84%, 平均76%、73%、72%和77%(气象数据取自网站http://www.wunderground. com)。10月采样期间后三天(23~25日)有一明显的降温过程, 温度由之前的24 ℃降至16~17 ℃; 1月采样过程中前三天(3~5日)气温为6~7 ℃, 相对湿度大于80%, 而8~10日温度降至-1~2 ℃, 相对湿度也降至51%~68%。
采样系统包含2.5 μm粒径切割装置, 进气口后部连接了两种不同性质的哈弗蜂窝式溶蚀器(Harvard honeycomb denuder), 一种用于吸附酸性气体组分如HNO3、HCl和SO2等, 另一种用于吸附碱性气体如NH3, PM2.5颗粒物用特氟龙(Teflon)滤膜(R2PJ047, 2.0 μm孔径, Gelman Science, 美国)采集, 后加两层后置膜, 一层为尼龙膜(Nylon), 用于吸附HNO3和HCl气体, 另一层为柠檬酸涂布的石英膜, 用以吸收NH3, 采样流速为10 L/min。与此同时, 另一采样器与溶蚀器/后置膜采样系统并排放置, 但按常规方法只用Teflon膜采集PM2.5, 通过两个采样器的比较以分析常规膜采样的误差。
溶蚀器和柠檬酸涂布石英膜的准备参照USEPA的推荐方法进行[7], 将第一个干净溶蚀器用含有1%Na2CO3与1%甘油的50%H2O+50%甲醇混合溶液涂布, 涂布后的溶蚀器放于真空干燥器内干燥; 第二个溶蚀器用含有4%柠檬酸与2%甘油的甲醇溶液涂布, 涂布后也放入真空干燥器内干燥。石英膜用含有4%柠檬酸与2%甘油的甲醇溶液涂布。溶蚀器和滤膜从干燥器中取出后立即装入采样器进行样品采集。
1.2 离子分析
采样结束后立即对溶蚀器和样品膜进行分析。其中滤膜直接放入干净聚丙烯塑料瓶中, 加入10 mL的超纯水(Mill-Q, 电阻率>18M), 室温超声抽提20 min, 抽提液经0.45 μm过滤膜过滤后进行离子色谱测定。溶蚀器用15 mL超纯水浸泡震荡30 min后倒出抽提液过滤后进行离子色谱分析。
离子色谱分析过程中采用外标法进行定量, 通过对0.5、1、2、5和10 μg/g 5种浓度标准溶液的离子色谱分析建立目标离子的工作曲线。
1.3 质量保证与质量控制(QA/QC)
溶蚀器和滤膜涂布、干燥过程中通过在真空干燥器中通入高纯氮以阻止其与空气的直接接触, 空白实验结果显示目标离子的空白值小于样品浓度的5%, 能满足实验要求。重复实验表明离子分析的相对偏差小于5%。
采用基质加标法分析了实验的回收率: 选择部分样品分成相同面积的两份, 其中一份用超纯水室温超声抽提后直接用离子色谱仪测定各目标离子的浓度, 另一份中加入2 μg/g的阴、阳离子标准溶液后再进行超声抽提, 通过计算标样加入前后各离子浓度得到离子的回收率。结果表明各目标离子的回收率都达到92%以上。
2 结果与讨论
2.1 上海PM2.5中主要水溶性离子浓度与组成
溶蚀器/后置膜采样实验中Teflon膜采集的颗粒物中各离子的浓度以及后置膜中各离子的浓度列于表1。
由于溶蚀器系统除去了大气中存在的酸性和碱性气体, 后置膜中测得的离子主要来自于采集于Teflon膜的颗粒物在样品采集过程中的挥发以及由于颗粒-颗粒之间发生相互反应而导致的挥发作用。因此, 大气PM2.5中各离子的真实浓度为Teflon膜与后置膜中浓度的和(表2中amb列)。硫酸/硫酸盐的挥发性非常小, 后置膜中基本检测不到硫酸根离子。
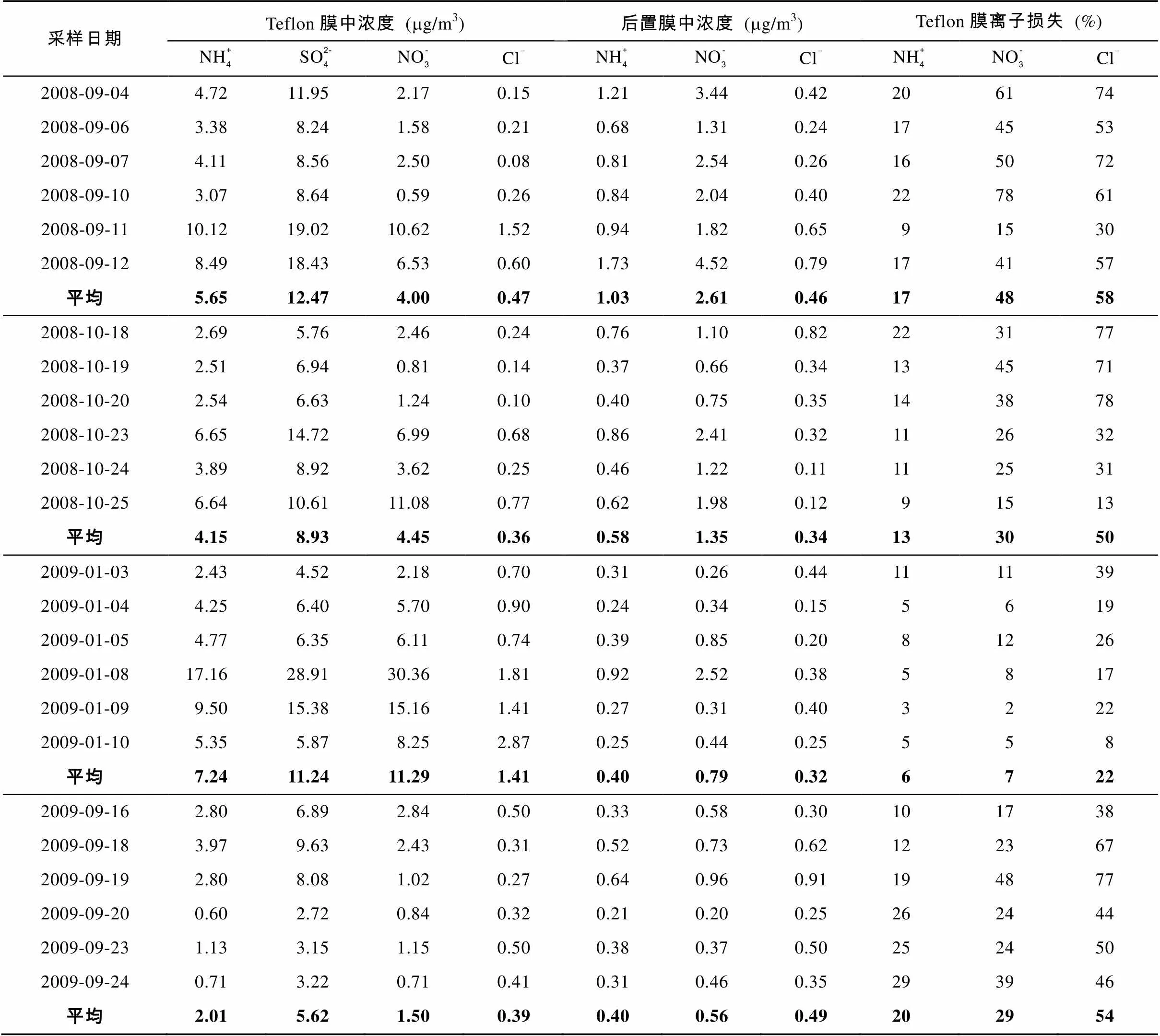
表1 溶蚀器/后置膜采样系统中主要离子浓度及膜损失
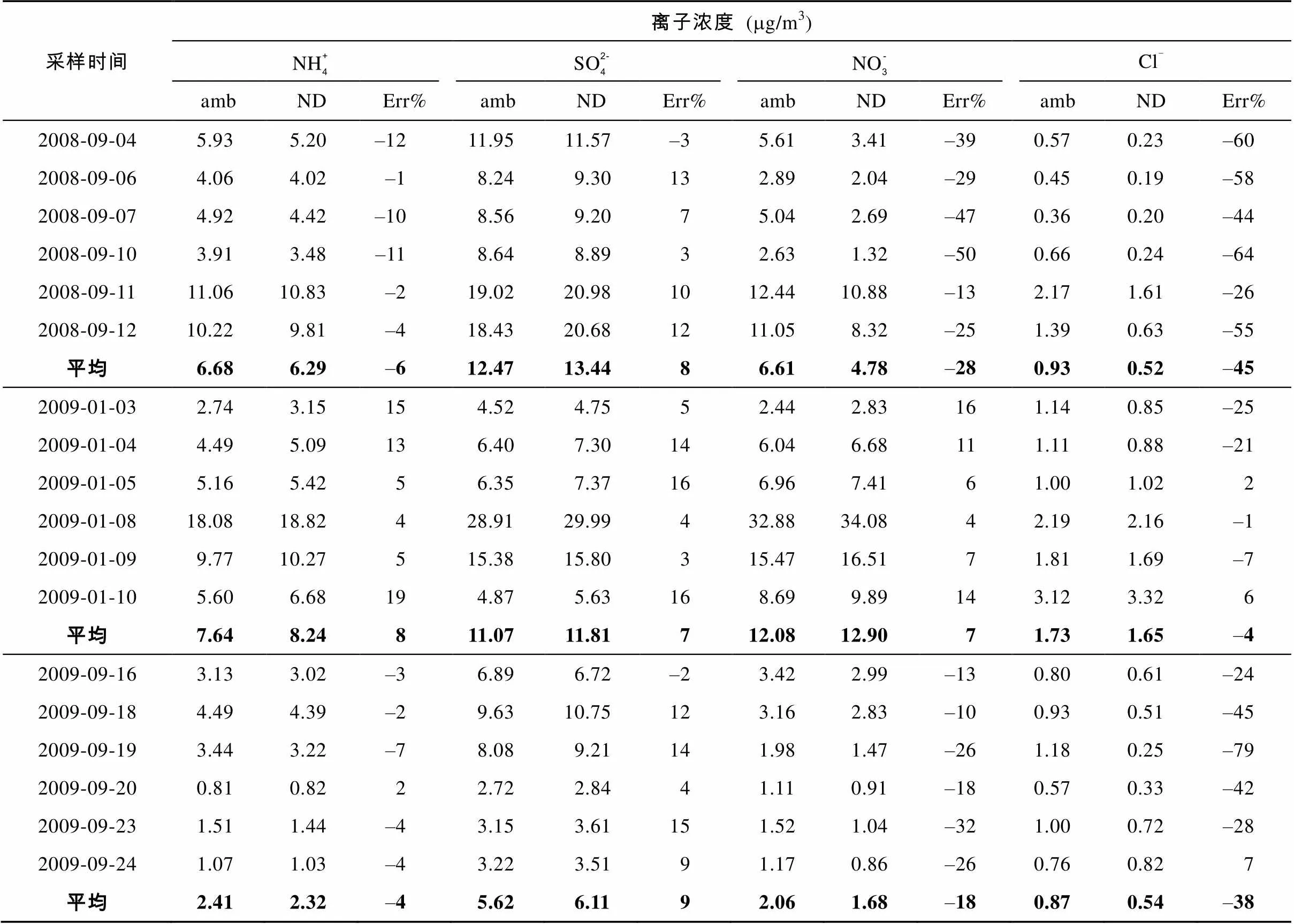
表2 无溶蚀器采样时PM2.5中主要离子的浓度及采样误差
注: amb表示带溶蚀器采样时Teflon膜和后置膜的总浓度; ND 表示不带溶蚀器的常规采样时Teflon膜中浓度; Err%表示常规采样中的误差百分比。
由表1可以看出, 硫酸根是4个定量的离子中最主要的离子, 在2008年9月、10月和2009年1月、9月4个采样期间的平均浓度分别为12.47、8.93、11.24和5.62mg/m3, 其次为硝酸根和铵根离子, 而氯离子浓度相对较低, 这几种离子的浓度与分布情况与已有关于上海PM2.5的报道值相近[8–9]。2009年9月采样期间浓度较低的一个原因是扩散条件较好, 来自海上的干净空气稀释了当地的污染物; 同时, 9月20日样品采集期间发生了一次明显的降雨过程, 导致了污染物浓度的明显降低。与硫酸根和铵根离子相比, 硝酸根离子浓度具有更明显的季节变化, 1月采样期间的浓度显著高于9、10月份, 说明了硝酸铵的挥发性对其浓度的巨大影响。
铵根、硝酸根和硫酸根的总浓度(ANS)在4个采样期的平均浓度为25.77、19.38、30.98和10.08mg/m3, 所有样品的平均总浓度为21.55mg/m3。Ye.[8]对上海PM2.5的研究表明, ANS浓度占PM2.5质量浓度的40%~45%, 则由本次实验的数据推测, 上海2008年至2009年PM2.5质量浓度在50mg/m3左右, 与上海市环保局公布的年均浓度基本一致, 说明上海的PM2.5浓度仍明显高于我国新的PM2.5空气质量标准(35mg/m3)。
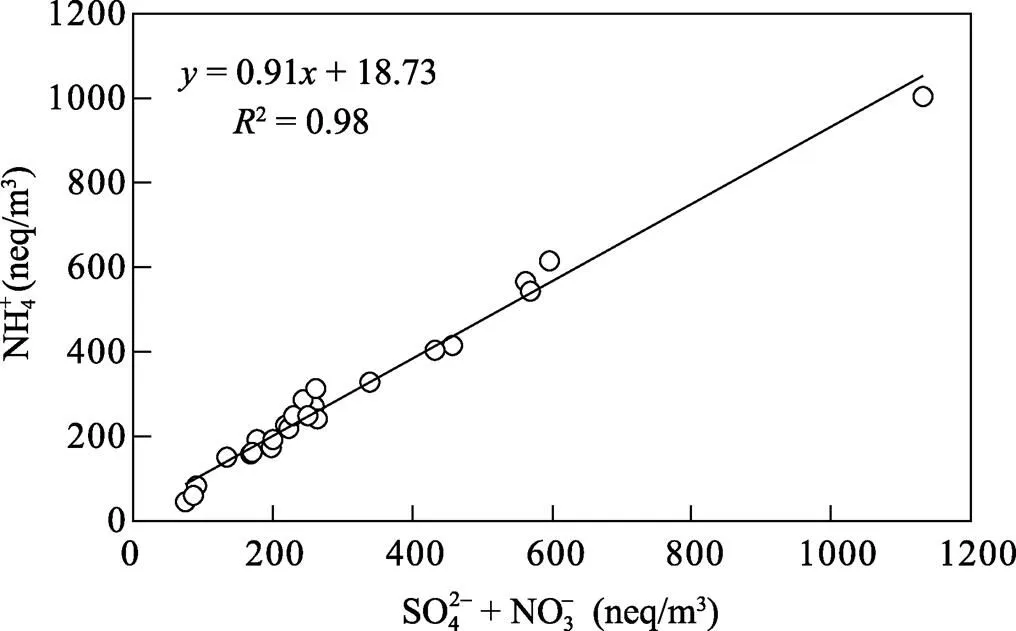
图1 上海PM2.5中铵根、硫酸根和硝酸根当量浓度之间的相关性
由于溶蚀器的使用会一定程度地破坏颗粒物中半挥发性组分与大气间的平衡, 从而会加大采样过程中颗粒态半挥发性离子的损失, 因此, 应用溶蚀器去除气体对颗粒物采样的影响时必须增加后置膜以校正挥发损失。
2.2 常规PM2.5采样中主要水溶性离子的采样误差分析
虽然已经发展了许多在线大气颗粒物监测技术, 但在PM2.5颗粒物化学组成的研究中最常见的采样方法还是通过膜过滤法采集样品然后在实验室进行分析, 样品采集时间一般持续12 h以上; 又由于溶蚀器/后置膜采样系统操作比较繁琐, 采样流速也有较大限制, 常规采样工作中通常只用单层滤膜。因此, 在常规颗粒物采样过程中, 收集到膜上的颗粒物除可能发生的颗粒-颗粒之间的相互作用及挥发损失外, 颗粒物与气体之间可能发生反应, 从而使采集到的颗粒物的组成发生变化, 导致采样误差的产生。
除前面讨论的颗粒-颗粒相互作用及平衡破坏导致的挥发损失外, 常规采样过程中空气中的气态组分穿过滤膜时可能被采样膜及膜上颗粒物吸附并与颗粒物组分发生反应, 造成颗粒物组成的改变。例如, 采样膜及已采集的颗粒物吸附空气中的氨气会中和颗粒中原有的酸性组分, 同时增加颗粒物中铵根离子的含量; 硝酸气体的吸附会增加颗粒中硝酸根的浓度, 还可能导致颗粒物中原有氯离子的损失。本次实验1月(冬季)采样期间铵根、硝酸根离子出现正误差, 说明气体吸咐是主要的误差来源, 冬季气温较低, 半挥发性组分的挥发性显著减小, 所以冬季采样时挥发导致的误差基本可以忽略。
硫酸盐及硫酸的沸点较高, 在常见大气条件下不容易发生挥发现象, 所以硫酸根离子的采样误差主要来源于采样膜以及膜上已采集的颗粒物对二氧化硫气体的吸收; SO2被吸附到膜及膜上的颗粒物后经过氧化反应形成硫酸根。根据Coutant的资料[16], 采样石英膜可以吸收环境中约5%的二氧化硫, 玻璃膜由于其碱性明显强于石英膜, 因而对二氧化硫的吸附更强[17]。Tsai.[18]研究发现, 在铵根离子比较充足情况下, 使用石英膜采集颗粒物时硫酸根离子的平均采样误差为11%。由于Teflon膜呈中性且具有较强的惰性, 通常认为Teflon膜对SO2的吸附作用可以忽略不计, 采样误差主要来源于已采集颗粒物对SO2的吸附。Pathak.[19]发现在氨比较充足的情况下, 颗粒物吸附SO2产生的硫酸根测量误差约为7%, 与本次实验的结果基本一致。
2.3 大气中气态NH3、HNO3和HCl浓度
通过溶蚀器的吸收作用可以测定大气中酸性及碱性气体如HNO3、HCl和NH3的浓度, 本次实验的结果列于表3。
由表3可以看到, 上海大气中NH3有较高的浓度(1.56~20.84 µg/m3), 2008年9月、10月和2009年1月、9月4次采样期间的平均浓度分别达到12.54、13.33、5.26和4.16 µg/m3, 所有样品的平均浓度为8.78 µg/m3; HNO3气体在各采样期的平均浓度小于2 µg/m3, 而HCl的浓度更低, 平均小于1 µg/m3。本次实验的NH3浓度与珠三角的测量结果(10~11月, 7.3 µg/m3)相比略高, 而HNO3和HCl的浓度低于珠三角但高于美国北卡罗来纳州的测量结果[20–21]。从表3还可以看出, 这3种气态组分的浓度存在一定的季节变化, 冬季浓度较低。由于HNO3和HCl具有半挥发性, 可以在气态和颗粒态两相间分配, 冬季较低的浓度可能是由于其更多地存在于颗粒态中。HNO3主要来源于大气光化学过程[20], 冬季较低温度下较低的生成速率也是导致低浓度的原因之一。NH3主要来源于畜禽、化肥、生物质燃烧及机动车尾气等一次排放[22–23], 冬季较低的浓度主要缘于地表挥发量的减少。
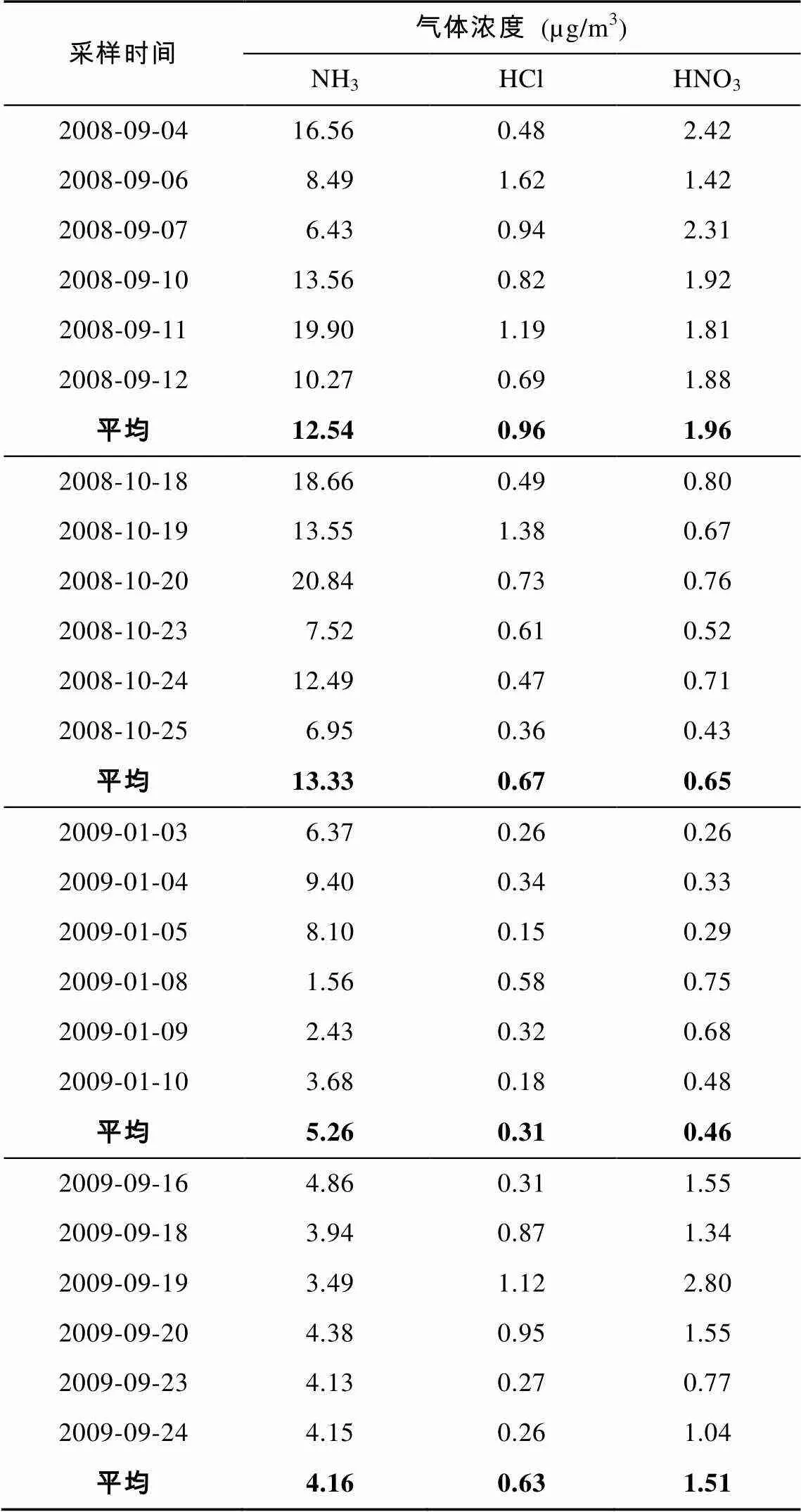
表3 溶蚀器测量的上海大气中酸、碱性气体的浓度
3 主要结论
(1)上海PM2.5中硫酸根、硝酸根和铵根离子总浓度的平均值超过20 µg/m3, 依然处于很高的水平, 机动车尾气已成为主要的污染源。
(3)上海大气中存在较高浓度的氨, 各采样期的平均浓度为8.78 µg/m3; HNO3气体的平均浓度小于2 µg/m3, HCl的平均浓度小于1 µg/m3, HNO3和HCl浓度具有显著的夏高冬低的季节性变化特征。
[1] Pope III C A, Burnett R T, Thun M J, Calle E E, Krewski D, Ito K, Thurston G D. Lung cancer, cardiopulmonary mortality, and long-term exposure to fine particulate air pollution [J]. J Am Med Assoc, 2002, 287(9): 1132–1141.
[2] Sloane C S, Watson J G, Chow J, Pritchett L, Richards L W. Size-segregated fine Particle measurements by chemical species and their impact on visibility impairment in Denver [J]. Atmos Environ, 1991, 25(5/6): 1013–1024.
[3] 宋玉芝, 秦伯强, 杨龙元, 胡维平, 罗潋葱. 大气湿沉降向太湖水生生态系统输送氮的初步估算 [J]. 湖泊科学 2005, 17(3): 226–230. Song Yu-zhi, Qin Bo-qiang, Yang Long-yuan, Hu Wei-ping, Luo Lian-cong. Primary estimation of atmospheric wet deposition of nitrogen to aquatic ecosystem of LakeTaihu [J]. J Lake Sci, 2005, 17(3): 226–230 (in Chinese with English abstract).
[4] Trebs I, Meixner F X, Slanina J, Otjes R, Jongejan P, Andreae M O. Real-time measurements of ammonia, acidic trace gases and water-soluble inorganic aerosol species at a rural site in the Amazon Basin[J]. Atmos Chem Phys, 2004, 4: 967–987.
[5] Yao X H, Shairsingh K, Lam P H, Evans G J. Underestimation of sulfate concentration in PM2.5using a semi-continuous particle instrument based on ion chromatography [J]. J Environ Monit, 2009, 11(6): 1292–1297.
[6] Pathak R K, Yao X H, Chan C K. Sampling artifacts of acidity and ionic species in PM2.5[J]. Environ Sci Technol, 2004, 38(1): 254–259.
[7] USEPA, Determination of strong acidity of atmospheric fine-particles (<2.5mm) using annular denuder technology [R]. EPA Report No. EPA/600/R-93/037; Atmospheric Research and Exposure Assessment Laboratory: Washington, DC, 1992.
[8] Ye B M, Ji X L, Yang H Z, Yao X H, Chan C K, Cadle S H, Chan T, Mulawa P A. Concentration and chemical composition of PM2.5in Shanghai for a 1-year period[J]. Atmos Environ, 2003, 37(4): 499–510.
[9] Wang Y, Zhuang G S, Zhang X Y, Huang K, Xu C, Tang A H, Chen J M, An Z S. The ion chemistry, seasonal cycle, and sources of PM2.5and TSP aerosol in Shanghai[J]. Atmos Environ, 2006, 40(16): 2935–2952.
[10] Ziemba L D, Fischer E, Griffin R J, Talbot R W. Aerosol acidity in rural New England: Temporal trends and source region analysis [J]. J Geophys Res, 2007, 112, D10S22, doi:10.1029/2006JD007605.
[11] Yao X H, Chan C K, Fang M, Cadle S, Chan T, Mulawa P, He K B, Ye B M. The water-soluble ionic composition of PM2.5in Shanghai and Beijing, China [J]. Atmos Environ, 2002, 36(26): 4223–4234.
[12] Xiao H, Liu C. Chemical characteristics of water soluble components in TSP over Guiyang, SW China, 2003[J]. Atmos Environ, 2004, 38(37): 6297–6306.
[13] Koutrakis P, Thompson K M, Wolfson J M, Spengler J D, Keeler G J, Slater J L. Determination of aerosol strong acidity losses due to interactions of collected particles: Results from laboratory and field studies [J]. Atmos Environ, 1992, 26(6): 987–995.
[14] Pathak R K, Yao X H, Alexsis K H L, Chan C K. Acidity and concentration of ionic species of PM2.5in Hong Kong [J]. Atmos Environ, 2003, 37(8): 1113–1124.
[15] Cheng Y H, Tsai C J. Evaporation loss of ammonium nitrate particles during filter sampling [J]. J Aerosol Sci, 1997, 28(8): 1553–1567.
[16] Coutant R W. Effect of environmental variables on collection of atmospheric sulfate[J]. Environ Sci Technol, 1977, 11(9): 873–878.
[17] Appel B R, Tokiwa Y, Haik M, Kothny E L. Artifact particulate sulfate and nitrate formation on filter media[J]. Atmos Environ, 1984, 18(2): 409–416.
[18] Tsai C J, Perng S N. Artifacts of ionic species for Hi-Vol PM10and PM10dichotomous samplers [J]. Atmos Environ, 1998, 32(9): 1605–1613.
[19] Pathak R K, Chan C K. Inter-particle and gas–particle interactions in sampling artifacts of PM2.5[J]. Atmos Environ, 2005, 39(9): 1597–1607.
[20] Hu M, Wu Z J, Slanina J, Lin P, Liu S, Zen L M. Acidic gases, ammonia and water-soluble ions in PM2.5at a coastal site in the Pearl River Delta, China[J]. Atmos Environ, 2008, 42(25): 6310–6320.
[21] Walker J T, Robarge W P, Shendrikar A, Kimball H. Inorganic PM2.5at a US agricultural site [J]. Environ Pollut, 2006, 139(2): 258–271.
[22] Krupa S V. Effects of atmospheric ammonia (NH3) on terrestrial vegetation: A review[J]. Environ Pollut, 2003, 124(2): 179–221.
[23] Huai T, Durbin T D, Younglove T, Scora G, Barth M, Norbeck J M. Vehicle specific power approach to estimating on-road NH3emissions from light-duty vehicles[J]. Environ Sci Technol, 2005, 39(24): 9595–9600.
[24] Zhuang H, Chan C K, Fang M, Wexler A S. Formation of nitrate and non-sea-salt sulfate on coarse particles [J]. Atmos Environ, 1999, 33(26): 4223–4233.
Concentrations and sampling artifacts of water-soluble ions in PM2.5in Shanghai sampled using denuder/backup-filter system
ZHANG Pan, ZHONG Mian, GUAN Jing-jing, JING Liang, LI Man, GONG Shi-yi and FENG Jia-liang*
(Institute of Environmental Pollution and Health, School of Environmental and Chemical Engineering, Shanghai University, Shanghai 200444, China)
PM2.5; water-soluble ions; sampling artifacts; ammonia; Shanghai
P593
A
0379-1726(2013)03-0197-08
2013-01-29;
2013-02-15;
2013-02-25
国家自然科学基金(20877052, 41173097)
张攀(1987–), 男, 硕士研究生, 环境工程专业。E-mail: p.z0810@163.com
FENG Jia-liang, E-mail: fengjialiang@shu.edu.cn, Tel: +86-21-66137738
