Ehlers-Danlos 综合征
2013-06-13薛旭红沈建雄
薛旭红,沈建雄
Ehlers-Danlos 综合征(Ehlers-Danlos Syndrome,EDS)又称弹力过度性皮肤伴皮肤和关节松弛的皮肤毛细管破裂,为真性结缔组织病,属于遗传性结构蛋白病。EDS 为罕见的结缔组织遗传疾病,当其累及骨关节系统将会有关节疼痛、肿胀及不稳、脊柱畸形等表现。最早由Tschernogobow 于1892 年报道,作者所描述的典型症状为皮肤弹性增高,关节过度活动以及骨关节假瘤样突起[1]。
目前国际上所公认的疾病名分别来自于丹麦的皮肤科医师Ehlers 及法国的皮肤科医师Danlos 分别在1901 年与1908 年的报道[2],他们指出这些缺陷是由于结缔组织异常所致,故命名为EDS。该病男女均可累及,男性发生率较高.往往有家族史,多数属常染色体显性遗传,在少数家庭本病是以性连锁隐性遗传的特征出现的。EDS 不同亚型遗传方式并不相同。根据遗传、临床表现及生化改变可将其分为11 种类型,其中Ⅵ型主要表现为皮肤弹性增大、关节过度活动,部分患者合并脊柱侧凸及眼球病变。该病十分罕见,发生率低于1/50 000。本院于2008 年2 月诊断并治疗了1 例以脊柱病变为主要表现的Ⅵ型EDS 患者。行侧凸矫正脊柱融合术,随访3 年。本文对此例病例资料进行总结并结合文献复习探讨EDS 合并脊柱侧凸的诊断及外科治疗策略。
1 病例资料
患者,女性,34 岁,因“发现背部不平10 年,加重2 年”入院。患者诉自幼皮肤松弛柔软,手、脚腕活动度大,能够过度背伸;磕碰等轻微外伤后容易出现血肿,全身轻微擦伤后容易生成瘢痕(见图1)。查体正常步态,颈部不短,正常体型。身高160 cm,体重55 kg,坐高92 cm,C7~S150 cm。躯干毛发正常,无蜘蛛指趾,腕征(-)。前额稍宽大,发际不低,巩膜及牙齿未见明显异常。全身皮肤松弛,弹性差,有关节过度活动表现;组织损伤后易出血,止血困难。除上述症状之外,无明显关节肿胀、疼痛,上肢精细动作正常。脊柱腰椎侧凸畸形,凸向左侧。双肩基本等高,枕外隆凸垂线自臀沟左侧0 cm通过,骨盆左低右高,高度差约1 cm。神经系统查体正常。
X 线片表现为:胸腰椎多个椎体形态异常(楔形变),胸弯Cobb 角30°,腰弯Cobb 角44°,伴有腰段后凸畸形,Cobb 角-50°。总体躯干平衡尚可,可见L1/L2有旋转半脱位的表现(见图2)。
2 诊 断
EDS 特征性表现为:①皮肤的弹性过度。②皮肤和血管十分脆弱,不破皮肤的轻微损伤,可以引起血肿或皮下出血[3]。创伤后留下香烟纸样的萎缩瘢痕,瘢痕后期可发生软疣样假性肿瘤。③关节的过度活动。④前额宽大,眼距、鼻梁较宽,并伴有先天性邹褶的内眦皮赘等特殊面容。⑤血管扩张性改变,如主动脉瘤、动脉夹层等,属于EDSⅣ型,是并发症最严重的一种类型[4]。⑥胫前和前臂的许多硬的豌豆大小的钙盐沉着结节。⑦EDS 常伴内脏损害。患者肠壁脆弱往往导致自发的肠破裂,反复的胃肠道出血和食管疝也可发生[5]。妊娠期子宫破裂常危及生命。前三条通常称为EDS 三联征:过度弹性皮肤、反复血肿和关节松弛。Pepin等[6]发现80%患者在40 岁以后有并发症发生,多数死于动脉破裂、肠道破裂或子宫破裂。其总结的81 例女性EDS 患者,其中有12 例死于妊娠期并发症。另外,少数患者可伴多发性神经纤维瘤病的典型表现。目前对EDS 的研究开始注重其亚型差异和临床多样性表现。1998 年英国医学遗传学家Beighton 根据临床表现分为6 型[7](见表1)。
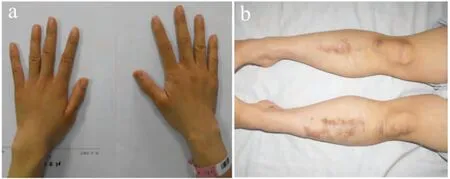
图1 术前大体图片Fig.1 Preoperative clinical pictures
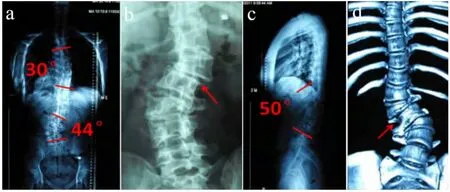
图2 术前影像学资料Fig.2 Preoperative radiologic data

表1 Ehlers-Danlos 综合征临床分型Tab.1 Classification and genetic causes of Ehlers-Danlos syndrom
临床诊断主要根据关节松弛,皮肤柔软及皮肤紫癍的存在。Ⅵ型与Ⅰ型的表现相似。但有脊柱后凸侧凸,Mafan 样体型及眼部病变,可以通过遗传史分析及临床检查给予诊断。主要的临床诊断指标包括:①皮肤张性增加,通常检查前臂掌侧的皮肤张性。②斑痕增宽萎缩,这是由伤口愈合不良及易于牵扯而导致的。③关节过度松弛,可以累及任何关节,9 个关节中至少5 个有松弛才算阳性。第5 指背伸>90°(2 个关节),拇指背伸接触前臂(2 个关节),肘关节过伸>10°(2 个关节),膝关节过伸>10°(2 个关节),腰部前屈能使手心接地(1 个关节)[4]。
本例患者诊断为Ⅵ型EDS。
3 治疗方法及结果
为防止侧凸继续发展,缓解疼痛症状,改善矢状面生理曲度,重建躯体平衡,行后路矫形内固定脊柱融合术,融合节段为T10~L5。术中取皮肤组织病理检查,结果提示慢性炎。术后腰弯改善为20°,腰段后凸改善为15°。
术后6 个月随访时,患者一般情况良好,疼痛缓解,畸形得到有效矫正,重建了矢状面平衡。术后2年随访,临床效果满意,患者诉腰部僵硬,其余无明显不适,脊柱正侧位X 线片示脊柱内固定位置良好,脊柱融合确实,融合远端无弯曲进展,躯干平衡良好,无失代偿现象(见图3)。
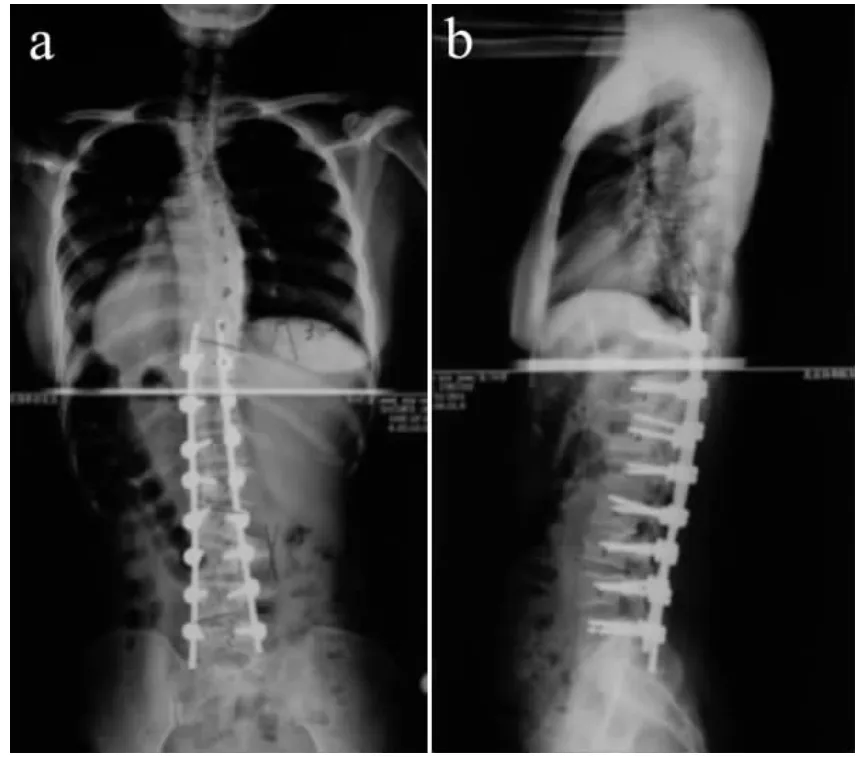
图3 术后影像学资料Fig.3 Postoperative radiologic data
4 讨 论
Ehlers-Danlos 综合征是一类少见的结缔组织遗传疾病,以皮肤弹性过度、关节过度伸展、组织脆性增加、易于损伤、创伤后不易愈合和无骨骼脆性增加为特征的一种胶原疾病[8]。该病是真皮的胶原纤维有缺陷,而弹力纤维正常,当其累及骨关节系统将会有关节疼痛、肿胀及不稳、脊柱畸形等表现。McKusick[9]对其主要临床表现归纳为皮肤光滑柔软、延展性增加,疤痕形成不良以及易留刮痕,关节过度活动及韧带组织脆性增加。除以上主要表现,次要表现包括血管及结肠破裂,眼睛与口腔并发症,血小板异常等。按照McKusick(1972)分类法分为11 型。本病呈遗传异质性,Ⅰ、Ⅱ、Ⅲ、Ⅳ、Ⅷ和Ⅺ型属常染色体显性遗传;Ⅳ(部分)、Ⅵ、Ⅶ、Ⅹ型为常染色体隐性遗传;Ⅴ、Ⅸ型呈X 连锁隐性遗传[9]。
在遗传的背景下,EDS 发生机制是结缔组织的缺陷,主要是胶原纤维量的缺陷及其形态上的异常,在真皮、皮下和关节囊里形成一种异常编织的疏松组织,而产生一系列临床症状。对于EDS 发生机理研究较多,多集中在动脉壁细胞外基质,如胶原、弹力蛋白的改变,血小板、毛细血管结构的变异以及酶学改变等方面。O’connell 等[10]研究发现键糖蛋白-X 为胶原纤维间的桥接分子,其缺失可能是EDS 发生的机制。Pepin 等[6]对一组EDS Ⅳ型患者及其亲属进行研究发现,患者的Ⅳ型前胶原基因(COL3A1)发生突变。但并发症类型与COL3A1 特定突变无关。Arteaga-Solis 等[11]认为EDS Ⅳ型的特点为大血管的破裂,而这种表现是由于Ⅲ型胶原突变所致。Yeowell 等[8]发现EDS Ⅶ型则是由于前胶原肽酶的缺陷所致,这种酶缺陷使胶原形成变得无序而薄弱。
EDS Ⅵ型是罕见的结缔组织常染色体隐性遗传病,特征性症状包括关节过度伸展,皮肤弹性增高及明显的脊柱侧凸,有时合并眼睛的临床表现及血管系统的异常。其根本生化缺陷是赖氨酸羟化酶基因的突变导致不同组织中赖氨酸羟化酶含量减低造成其活性的丧失。而赖氨酸羟化酶是胶原组织中交联结构形成的关键酶,该酶缺失直接导致结缔组织中胶原含量及结构异常,最终引起上述临床症状[12]。对Ⅵ型EDS 合并脊柱侧凸患者的治疗,仅有少数文献报道,其手术指征目前尚无统一标准[13]。有学者认为由于出血及软组织愈合不良等潜在风险,应尽量避免手术[14]。
Rabenhorst 等[15]报道了一组6 例Ehlers-Danlos综合征合并严重脊柱侧凸的患者,均行后路脊柱矫形内固定植骨融合术,共行14 例次手术。有8 例次出现并发症,包括血肿形成,内固定失败,切口感染及死亡。其认为对于此类患者,尽量采取相对保守的手术方法,术前充分评估,术中及术后需要格外注意出血及感染的发生。
Ⅵ型EDS 是一类极为罕见的先天性疾病,若脊柱侧凸持续加重且合并腰背疼痛等临床症状,应及时予以外科干预。术前应对患者全身情况进行评估,术中应控制性降压,仔细止血,适当应用止血药,术中仔细操作,手术相对安全。此类患者应行后路长节段固定融合,这样能有效避免弯曲进展及躯干失平衡。特别注意谨慎选择前路手术,避免大血管损伤破裂造成严重的并发症甚至死亡。
[1]Parapia LA,Jackson C.Ehlers-Danlos syndrome-a historical review[J].Br J Haematol,2008,141(1):32-35.
[2]Uitto J.The Ehlers-Danlos syndrome--phenotypic spectrum and molecular genetics[J].Eur J Dermatol,2005,15 (5):311-312.
[3]Blaszczyk M,Depaepe A,Nuytinck L,et al.Acrogeria of the Gottron type in a mother and son[J].Eur J Dermatol,2000,10(1):36-40.
[4]Shirley ED,Demaio M,Bodurtha J.Ehlers-danlos syndrome in orthopaedics:etiology,diagnosis,and treatment implications[J].Sports Health,2012 ,4(5):394-403.
[5]Malfait F,Wenstrup RJ,De Paepe A.Clinical and genetic aspects of Ehlers-Danlos syndrome,classic type[J].Genet Med,2010,12(10):597-605.
[6]Pepin M,Schwarze U,Superti-Furga A,et al.Clinical and genetic features of Ehlers-Danlos syndrome type IV,the vascular type[J].N Engl J Med,2000,342(10):673-680.
[7]Beighton P,De Paepe A,Steinmann B,et al.Ehlers-Danlos syndromes:revised nosology,Villefranche,1997.Ehlers-Danlos National Foundation (USA)and Ehlers-Danlos Support Group(UK)[J].Am J Med Genet,1998,77(1):31-37.
[8]Yeowell HN,Pinnell SR.The Ehlers-Danlos syndromes[J].Semin Dermatol,1993,12(3):229-240.
[9]McKusick VA.The Ehlers-Danlos syndrome[M].//Heritable Disorders of Connective Tissue.4th ed.St.Louis:Mosby,1972:292-371.
[10]O’Connell M,Burrows NP,van Vlijmen-Willems MJ,et al.Tenascin-X deficiency and Ehlers-Danlos syndrome:a case report and review of the literature[J].Br J Dermatol,2010,163(6):1340-1345.
[11]Arteaga-Solis E,Gayraud B,Ramirez F.Elastic and collagenous networks in vascular diseases[J].Cell Struct Funct,2000,25(2):69-72.
[12]De Paepe A,Malfait F.The Ehlers-Danlos syndrome,a disorder with many faces[J].Clin Genet.2012,82(1):1-11.
[13]Jasiewicz B,Potaczek T,Tesiorowski M,Lokas K.Spine deformities in patients with Ehlers-Danlos syndrome,type IV- late results of surgical treatment[J].Scoliosis,2010,5:26.
[14]Liu Y,Gao R,Zhou X,et al.Posterior spinal fusion for scoliosis in Ehlers-Danlos syndrome,kyphoscoliosis type[J].Orthopedics,2011,34(6):228.
[15]Rabenhorst BM,Garg S,Herring JA.Posterior spinal fusion in patients with Ehlers-Danlos syndrome:a report of six cases[J].J Child Orthop,2012,6(2):131-136.
