有机标准物质期间核查核磁共振定量分析仪器参数的研究*
2013-06-05韦英亮潘艳坤杨益林
韦英亮,潘艳坤,杨益林
(广西分析测试研究中心,南宁 530022)
有机标准物质期间核查核磁共振定量分析仪器参数的研究*
韦英亮,潘艳坤,杨益林
(广西分析测试研究中心,南宁 530022)
以磺胺对甲氧嘧啶为例,对有机标准物质期间核查核磁共振定量分析方法的仪器参数作了研究,主要包括脉冲宽度、弛豫延迟时间、扫描次数、谱宽、发射机频率偏移、时域数据长度。分别对磺胺、磺胺对甲氧嘧啶、磺胺间甲氧嘧啶、磺胺噻唑、磺胺嘧啶、磺胺吡啶、磺胺苯吡唑7种磺胺类标准物质进行期间核查含量测定,测定结果的相对误差为0.1%~0.3%,准确度与高压液相色谱方法相当。
有机标准物质;期间核查;核磁共振定量分析;磺胺对甲氧嘧啶;磺胺类标准物质
核磁共振(Nuclear Magnetic Resonance,NMR)光谱发现于1945年,虽然上世纪70年代就有人提出核磁共振谱可用于定量分析,但由于仪器灵敏度低,重现性差,测定所需样品量大,不能满足定量分析的要求,长期以来核磁共振波谱仪只是作为结构测定的工具。而超导傅里叶变换核磁共振波谱仪的出现使得仪器性能大为改善,定量分析的精密度、重现性和准确度达到或接近了高效液相色谱仪的水平[1]。
标准物质在时间上具有保存量值、在空间上具有量值溯源的功能。标准物质期间核查是验证标准物质是否处于校准状态,以确保分析结果的质量[2]。因此标准物质期间核查必须同时考察定性和定量两方面。
目前有机标准物质期间核查溯源性定值方法多为气相色谱法和液相色谱法[3],两者均需重新购买新的标准物质,或者用一级标准物质对二级标准物质进行校准,有时还必须进行工作曲线校准才可以得到比较可信的结果。测试过程中仪器精密度受温度、压力、流动相等因素影响大,若要确保保留时间和峰面积的较好稳定性就需要较长的仪器平衡时间。有的前处理需要衍生化等步骤,测试过程较为复杂耗时。核磁共振定性鉴定和定量分析可同步完成,定性鉴定无须标准样品,用于定性和定量时氢谱1H NMR的检测时间短,重复性好,无须对样品和内标物进行特别的前处理,只要溶于合适的氘代试剂即可测定。核磁共振波谱定量分析在我国已见诸报道[4-8],有机标准物质纯度满足核磁共振定量检测要求,而核磁共振定量分析用于有机标准物质期间核查溯源性定值的报道尚不多见。笔者选用磺胺对甲氧嘧啶作为定量分析研究的对象,以对硝基甲苯作为内标物,共含有4种不同弛豫机理的质子基团:甲基、芳氢、仲胺、叔胺。主要研究脉冲序列参数(脉冲宽度、延迟时间、扫描次数)及采样参数(发射机频率偏移、采样点数)等因素对上述质子峰面积的影响,探索最佳的定量分析仪器条件。
1 实验部分
1.1 主要仪器与试剂
核磁共振波谱仪:Bruker AvanceⅢ600型,5 mm BBO(宽带观测)探头,1H的观察频率为600.17 MHz;
分析电子天平:Shimadzu AUW220D型,感量为0.01 mg,日本岛津公司;
氘代二甲基亚砜(DMSO-d6):纯度为99.9%,上海谱振生物科技有限公司;
磺胺对甲氧嘧啶标准样品:纯度为99.4%,国家标准物质研究中心;
对硝基甲苯标准样品:纯度为99.5%,国家标准物质研究中心。
1.2 标准样品和内标溶液的制备
准确称取磺胺对甲氧嘧啶标准样品135.40 mg,置于5 mL容量瓶中,用氘代二甲基亚砜溶解并定容至标线,制成27.08 mg/mL磺胺对甲氧嘧啶标准物质溶液。
取内标物对硝基甲苯56.77 mg,置于2 mL容量瓶中,同上法制成28.38 mg/mL内标溶液。
1.3 计算公式
样品质量分数X的计算公式如下:
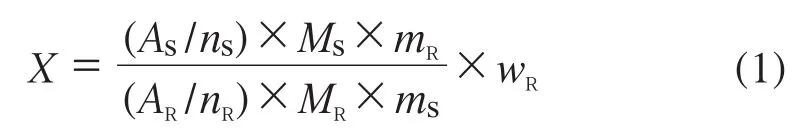
式中:AS——被测样品选定的积分面积;
ns——样品被积分信号包含的质子数;
Ms——被测样品的相对分子质量;
mR——称取的内标物质量;
wR——内标物的质量分数;
AR——内标物选定信号的积分面积;
nR——内标物被积分信号包含的质子数;
MR——内标物的相对分子质量;
ms——称取的样品质量。
1.4 仪器调节
将试样放入核磁共振谱仪中,调谐、匀场,然后测试氢谱,经过傅里叶变换,对所得的谱图进行相位校正、基线校正,确定结构正确无误后,分别对定量峰和内标峰进行积分。每个样品重复测定5次。
2 仪器参数条件的选择
2.1 脉冲宽度P1
实验参数设置如下:弛豫延迟时间D1=10 s;扫面次数为NS=8次;谱宽SW=15.019 7×10-6;发射机频率偏移为8.260×10-6;采样温度T=298.0 K;时域数据长度TD=64 K 。
样品:27.08 mg/mL磺胺对甲氧嘧啶(溶剂为DMSO-d6)。
信号积分处理:设磺胺对甲氧嘧啶芳环2位质子δ 7.60(2H,d,J=8.76 Hz)的积分面积为2.000 0,积分范围为δ 7.64~7.56;3位质子δ 6.57(2H,d,J=8.76 Hz)的积分范围为δ 7.60~7.52;3’位质子δ 8.26(2H,s)的积分范围为δ 8.30~8.22;伯胺根质子(—NH2) δ 5.96(2H,s)的积分范围为δ 5.99~5.91;仲胺根质子(—NH—) δ 10.99(1H,s)的积分范围为δ 11.02~10.94;甲氧基质子(—OCH3) δ 3.78(3H,s)的积分范围为δ 3.81~3.73。
脉冲的作用是建立横向磁化强度,1H NMR定量实验首先要求仪器的90°脉冲宽度必须准确,按照仪器脉冲校正的要求,采用0.1%乙基苯(溶剂为氘代氯仿)重新校正仪器氢谱的90°脉冲宽度,得到仪器90°脉冲宽度为14.25 μs。图1、图2为所测定磺胺对甲氧嘧啶及内标对硝基甲苯的核磁共振氢谱。
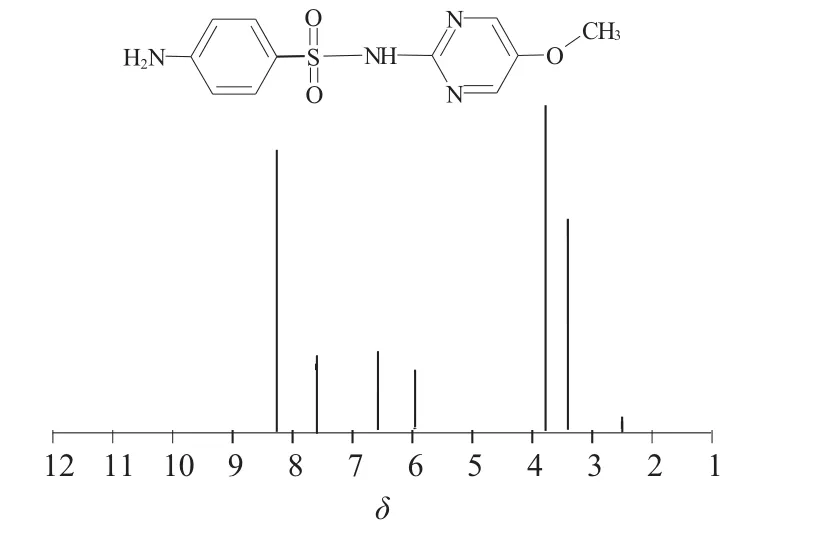
图1 磺胺对甲氧嘧啶的结构及其氢谱
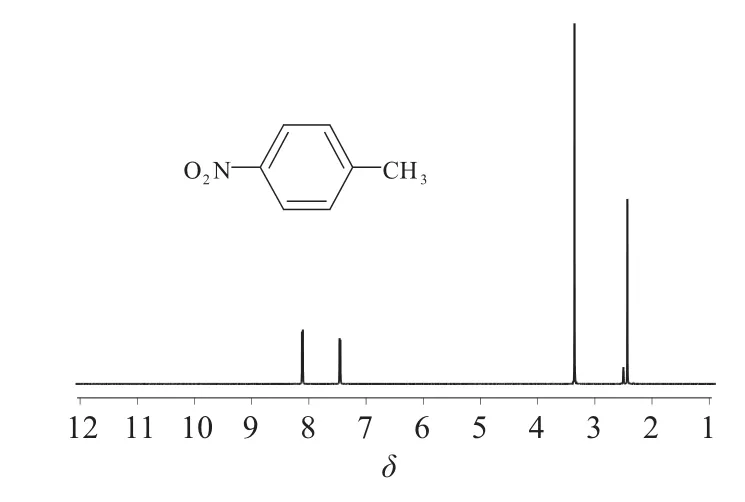
图2 对硝基甲苯的结构及其氢谱
从P1=1.00 μs开始,逐渐增加脉冲宽度,测得质子峰相对峰面积见表1。
由表1可见,随着P1的增加,芳香质子积分面积下降,活泼氢变化趋势不明显,甲基变化不大。当脉冲宽度小于5 μs时,只有芳香质子积分面积接近实际值2,甲基、活泼氢—NH2和—NH—都达不到实际值3,2和1。因为采用90°脉冲激发NMR样品以获取最强的发射信号,脉冲宽度增大后,需要增大激发脉冲之间的间隔时间使样品充分弛豫。如果需要重复激发样品,更有效的方法是使用30°脉冲激发,减少相应的弛豫时间。虽然每个放射信号比较弱,但更快的数据累加速度使30°脉冲在整体上拥有更好的灵敏度,所以脉冲宽度以不超过30°为宜。因此当仪器的90°脉冲宽度为14.25 μs时,其30°脉冲宽度应该是14.25 μs的三分之一,即为4.75 μs。与实际测定值小于5 μs一致。所以应该选择脉冲程序“zg30”,其唯一脉冲宽度是P1,即4.75 μs 。
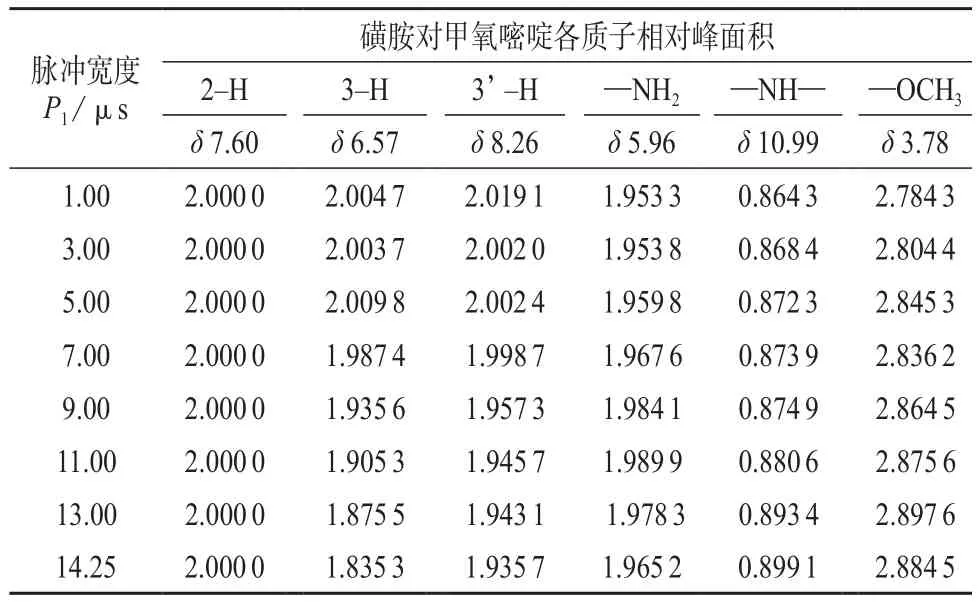
表1 脉冲宽度P1对相对峰面积积分的影响
实验选择磺胺对甲氧嘧啶2位质子δ 7.60(2H,d,J=8.76 Hz)作为定量峰,积分范围为δ 7.64~7.56;内标峰取对硝基甲苯中的δ 7.46(2H,d,J=8.40 Hz),积分范围为δ 7.50~7.42。
2.2 弛豫延迟时间D1
弛豫延迟时间D1对各质子峰相对峰面积影响的实验数据见表2。实验参数除延迟时间D1和P1=4.75 μs外,其它参数同2.1。
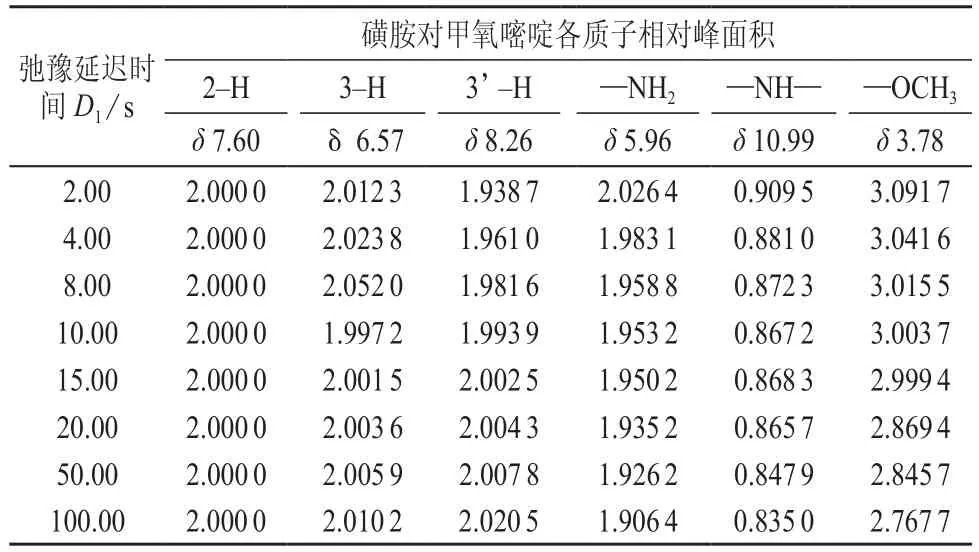
表2 延迟时间D1对各质子峰相对面积的影响
脉冲前延迟时间D1是为了保证实验之前系统有足够的宏观(纵向)磁化强度。一系列的脉冲和不足的弛豫时间激发NMR 样品会造成样品饱和。为了使样品释放出更多的能量,每个脉冲前都添加了“D1”延迟。这样可以使样品有足够的时间来弛豫。当选择脉冲程序“zg30”时,D1是唯一的延迟。由表2数据可以看出,不同环境的芳香质子随着延迟时间D1的增大相对峰面积有不同的变化趋势,但都是在延迟时间D1=10.00 s左右趋近实际值2 ,同时甲基也在在延迟时间D1=10.00 s左右趋近实际值3。而随着延迟时间D1的增大活泼氢相对峰面积有变小的趋势。因此可以确定实验的延迟时间D1=10.00 s。
2.3 扫描次数NS
实验了扫描次数NS对各质子峰相对面积的影响,结果见表3。实验参数除NS外,其它参数同2.1。
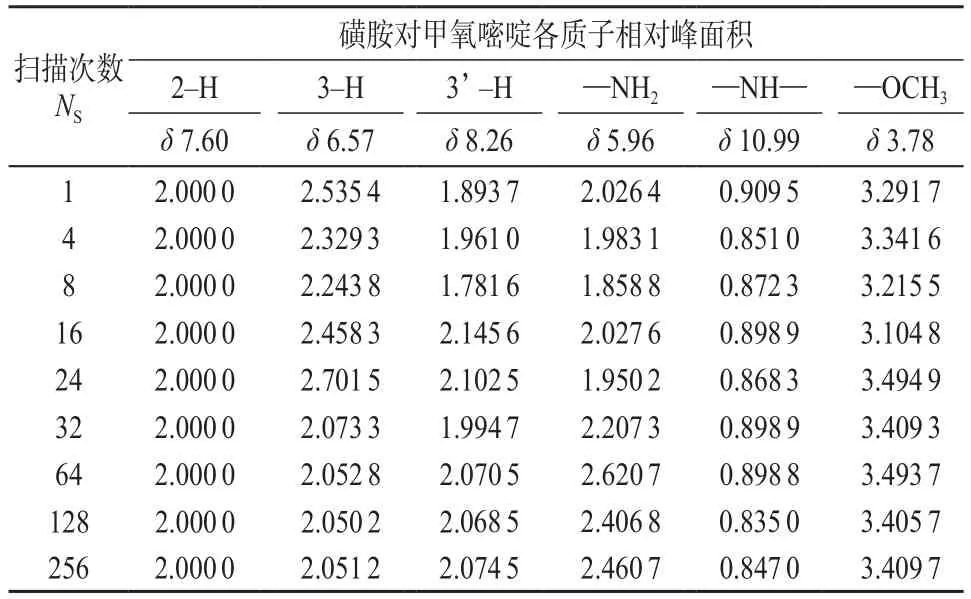
表3 扫描次数NS对各质子峰相对面积的影响
在NMR实验中,经常需要多次扫描来改善谱图质量,实验时间也相应增加。从表3中数据可以看出,对活泼氢和甲基质子来说,无论扫描次数多少,数据的重现性和稳定性都很差。扫描次数小于64次时,苯环质子的数据重现性也很差.只有当扫描次数不小于64次时,质子才趋近实际质子总数2,数据比较稳定,重现性好。可以确定扫描次数为64次即可满足一般分析的要求。这样处理是为了节省实验时间,提高分析效率。
2.4 发射机频率偏移
实验了发射机频率偏移对质子峰相对峰面积的影响,结果见表4。实验参数除脉冲宽度P1=4.75 μs,NS=64和发射机频率偏移外,其它参数同2.1。
之前设定的谱宽SW=15 ×10-6,从谱图上找到所有质子对应的信号。发射机频率偏移的确定是为了重新定义谱宽,在保证信号不丢失的情况下得到分辨率更高的谱图。从低场到高场,将发射机频率偏移分别设在每一个峰的中心位置上。表4数据显示,发射机频率偏移无论设在哪个点位上,苯环质子都接近实际值2,活泼氢质子—NH2和—NH—都分别小于2和1,甲基质子都大于3。为了得到分辨率较高的谱图,谱宽SW=15×10-6显然不合适,有必要减小谱宽,将谱宽设置为SW=13×10-6,发射机频率偏移设为5.96是合理的。
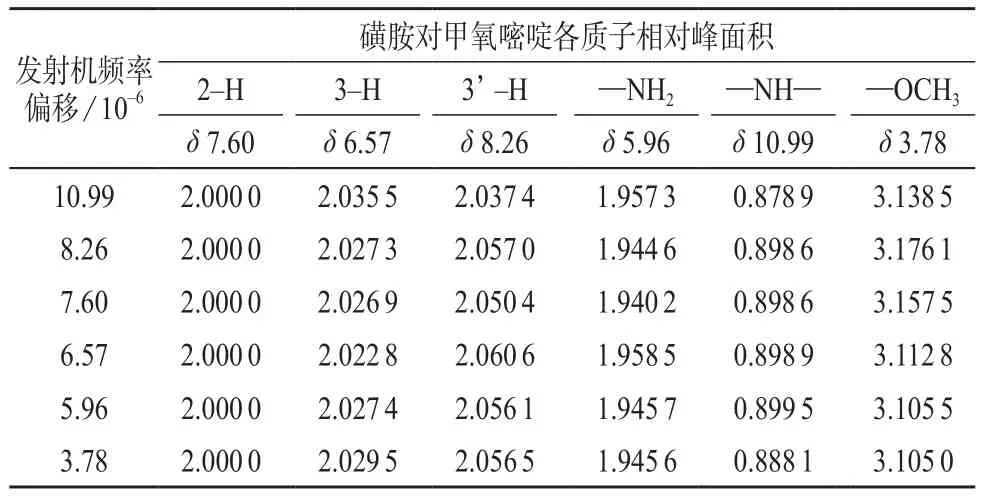
表4 发射机频率偏移对各质子峰相对面积的影响
2.5 其它参数
时域数据长度(Time Domain Data Size,TD)的值就是从FID中采样和数字化的点数(side of fid)。增大FID会改善FID 分辨率,同时也会需要更长的采样时间,但对峰面积影响不大,对1D FID,TD一般为16,32 或者64 K。对于定量实验取64 K。采样时间Acquisition Time(AQ),是完成一次扫描所需要的时间(单位为秒),根据TD 和SW计算得出。也可以直接修改AQ值,这时TD 的值会相应改变。总的来说,采样时间对质子峰相对峰积分面积没有明显影响。
3 结果与讨论
综合上述仪器条件,实验参数设置如下:90°脉冲宽度:14.25 μs;脉冲程序:zg30;脉冲延迟时间(D1):10.00 s;扫描次数(NS):64次;谱宽(SW):13 ×10-6;发射机频率偏移:5.69 ×10-6;时域数据长度(TD):64 K;采样温度(T):298 K。取磺胺对甲氧嘧啶标准物质溶液300.0 μL 和对硝基甲苯内标溶液300.0 μL,置于核磁管中,平行制备7份样品,测定磺胺对甲氧嘧相对含量,考察重复性结果见表5。由表5可知,方法的重复性良好。

表5 重复性试验结果 %
选择磺胺、磺胺对甲氧嘧啶、磺胺间甲氧嘧啶、磺胺噻唑、磺胺嘧啶、磺胺吡啶、磺胺苯吡唑7种标准物质,按上述仪器参数条件对每个样品的积分值测定3次,取其平均值,并与高压液相色谱检测结果比较,结果见表6。由表6可知,核磁共振法测定结果与高压液相色谱法的测定结果相近,相对误差均在0.1%~0.3%之间,满足磺胺类标准物质期间核查定值方法的要求。
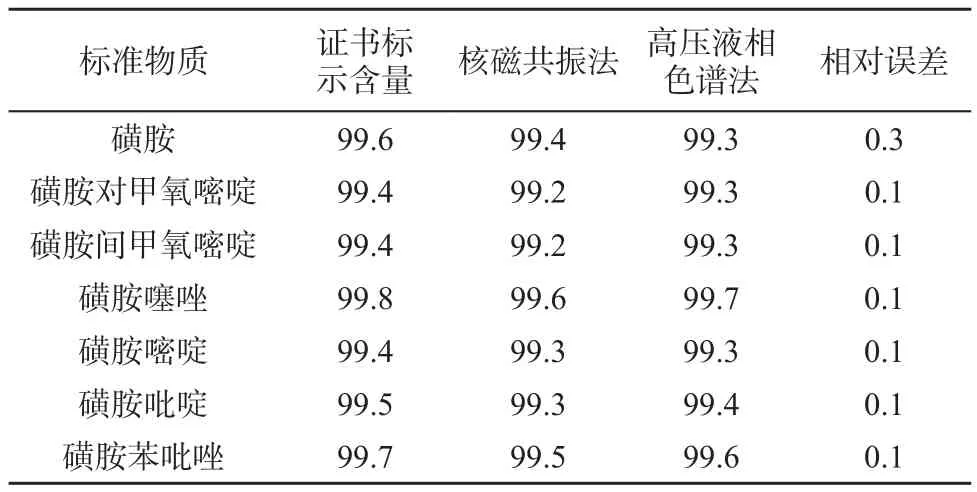
表6 磺胺类标准物质期间核查含量测定结果 %
综上所述,核磁共振定量分析应注意以下条件:
(1)确定准确的90°脉冲宽度,脉冲程序选择30°小角度脉冲;
(2)要有足够的弛豫延迟时间,保证实验之前系统有足够的宏观磁化强度;
(3)要有足够的扫描时间,保证谱图质量和各个质子积分面积的稳定性;
(4)设置合适的谱宽和发射机频率偏移,确保谱图有良好的分辨率;
(5)选择合理的定量峰和内标峰,甲基和活泼氢不适合作为定量峰;
(6)定量分析时,要认真匀场,磁场均匀才能保证谱仪的分辨率;相位校正可确保峰形左、右对称,呈正态分布的标准吸收峰形,为了避免出现边带应尽量不旋转样品;校正基线使所有质子的化学位移成水平直线;统一选用溶剂峰定标;先将谱图放大,取峰形轮廓线与完全水平的基线刚好重合处作为定量峰和内标峰的积分区域,这样才能够保证积分面积重现性良好。
4 结论
磺胺类有机物的基本结构为对氨基苯磺酰胺,采用对硝基甲苯为内标物可以对所有的磺胺类标准物质进行含量测定。因此,核磁共振核波谱法定性无须标准品,利用谱图化学位移、耦合常数、质子数可以给物质定性;只要选定一种合适的内标物,采用内标法,就可以对一系列结构相似的标准物质进行含量测定,为实验室节省大笔购买新标样的经费,节约实验成本。且定量分析稳定性好,只需测试一次就可以得到可靠的结果,分析效果与HPLC法相近。因此核磁共振波谱法可以作为实验室有机标准物质期间核查量值溯源性定值分析方法的辅助手段之一。
[1]Maniara G,Rajamoorthik K,Rajan S,et a1.Method performance and validation for quantitative analysis by1H and31PNMR spectroscopy. Applications to Analytical Standards and Agricultural Chemicals [J]. Anal Chem,1998,70: 4 921-4 928.
[2]马冲先.标准物质和实验室认可[J].理化检验:化学分册,2005,4(12): 947-951.
[3]韦英亮,潘艳坤.标准物质期间核查溯源性定值分析方法[J].化学分析计量,2012(5): 101-103.
[4]胡敏,胡昌勤,刘文英.核磁共振波谱法测定药物基准物质的绝对含量[J].分析化学,2004,32(4): 451-455.
[5]张伟,黄挺,徐蓓,等.苏丹红Ⅰ、Ⅱ标准物质纯度的核磁共振法测定[J].分析测试学报,2010,29(2): 194-198.
[6]卢蕾,李秀琴,邵春发,等. 5-甲基吗啡-3-氨基-2-唑烷基酮标准物质的研制[J].化学分析计量,2011,20(2): 4-8.
[7]邹明强,刘丽,王飞,等.自制异噁草酮标准物质纯度、均匀性及稳定性测定[J].化学试剂,2004,26(2): 91-92,104.
[8]毛希安.现代核磁共振实用技术及应用[M].北京:科学技术文献出版社,2000: 225-226.
Instrument Parameters of NMR for Quantitative Analysis of the Organic Standard Substances Period Verification
Wei Yingliang,Pan Yankun,Yang Yilin
(Guangxi Analytical Center,Nanning 530022,China)
Sulfameter as a sample, the instrument parameters of NMR including pulse width,delay time, number of scans, spectral width etc. for quantitative analysis to the organic standard substances period verification were discussed. The relative error of sulfanilamide, sulfameter, sulfamonomethoxine, sulfathiazloe, sulfadiazine, sulfapyridine and sulfaphenazolum reference materials determined by NMR were 0.1%-0.3%, which matched HPLC method in accuracy.
organic standard substance; period verification; NMR for quantitative analysis; sulfameter; sulfonamides reference Material
O657.61
A
1008-6145(2013)02-0070-05
10.3969/j.issn.1008-6145.2013.02.020
*广西壮族自治区直属公益性科研院所基本科研业务费专项(No.2011AC202)
联系人:韦英亮; E-mail: wyl3903895@163.com
2012-12-26
