黄芪药材、饮片中黄芪甲苷含量测定方法的改进
2013-06-05丁如贤李一珺孙雪妹杨明华
丁如贤 李 霞 李一珺 孙雪妹 杨明华
(上海市浦东食品药品检验所,上海,201203)
黄芪药材、饮片中黄芪甲苷含量测定方法的改进
丁如贤 李 霞 李一珺 孙雪妹 杨明华
(上海市浦东食品药品检验所,上海,201203)
目的:建立操作简便、重现性好的黄芪药材、饮片中黄芪甲苷的提取及含量测定方法。方法:比较4种不同的提取方法,用HPLC-ELSD法测定黄芪甲苷含量,色谱柱为Capcell PAK C18(4.6 mm×250 mm,5 μm);流动相为乙腈-水(32∶68)。结果:简化的提取方法,黄芪甲苷的进样量在0.53~12.72 μg范围内线性关系良好(r=0.999 4),平均回收率为98.91%,RSD=2.10%。结论:改进方法样品提取简单、方便、准确、重现性好,可作为黄芪药材、饮片中黄芪甲苷含量测定方法。
黄芪;黄芪甲苷;含量测定;高效液相蒸发光检测
黄芪为常用中药,是豆科植物蒙古黄芪Astragalus membranaceus(Fisch.)Bge.var.mongholicus(Bge.)Hsiao或膜荚黄芪Astragalus membranaceus(Fisch.)Bge.的干燥根,具有补气固表,利尿托毒等功效。黄芪中主要成分为皂苷类、黄酮类、多糖类等,其中黄芪甲苷具有正性肌力保护血管内皮细胞、抗炎、抗病毒等多方面药理活性[1],是黄芪药材中的重要活性成分,通常作为黄芪药材及其制剂质量控制的主要指标。
中华人民共和国药典(以下简称《中国药典》)2010年版[2]采用HPLC-ELSD测定黄芪中黄芪甲苷的含量,样品前处理过程繁琐、耗时、影响因素多,直接导致方法的回收率较低、重现性较差,对处于合格边缘检测样品的准确判断有非常大的困难。有些文献[3-4]报道的方法与药典基本类似,定量检测的准确性较差。也有文献[5-6]报道对样品前处理进行简化,仅采用超声处理,然后用HPLC-ELSD梯度洗脱的方法测定药材中黄芪甲苷的含量,但检测结果也不理想。
我们建立了黄芪样品的处理方法,简化了样品的制备过程,缩短前处理时间和步骤,提高了测定的准确性、稳定性,现报道如下。
1 仪器与试药
Waters 2695高效液相色谱仪,Waters 2424 ELS检测器,Empower色谱数据工作站;BOLONG USC-702超声波清洗器;D101大孔吸附树脂柱(柱内径1.5 cm,长12 cm)。
黄芪饮片(上海虹桥中药饮片有限公司,批号:20110614);黄芪甲苷对照品(中国食品药品检定研究院,批号:110781-200613)。甲醇(色谱纯)、乙腈(色谱纯),冰醋酸(分析纯),水(超纯水)。
2 方法与结果
2.1 色谱条件 色谱柱:Capcell PAK C18(4.6 mm× 250 mm,5 μm);流动相:乙腈-水(32∶68);柱温:30℃;流速:1 mL/min;Waters 2424 ELS检测条件:增益60,漂移管温度65℃,氮气压力40Psi,雾化器温度39℃。
2.2 溶液的制备
2.1.1 对照品溶液制备 精密称取在放置五氧化二磷的减压干燥器中干燥24 h黄芪甲苷对照品10.60 mg,置20 mL量瓶中,加甲醇溶解,并稀释至刻度,摇匀,即得(每1 mL中含黄芪甲苷0.53 mg)。
2.2.2 供试品溶液制备 将黄芪饮片粉碎,备用。
方法(a)(2010版《中国药典》方法)供试品制备:取本品粉末约4 g,精密称定,置索氏提取器中,加甲醇40 mL,冷浸过夜,再加甲醇适量,加热回流4 h,提取液回收溶剂并浓缩至干,残渣加热水10 mL,超声溶解,转移至分液漏斗,用水饱和的正丁醇提4次,每次40 mL,合并正丁醇液,氨试液洗2次,每次40 mL,弃去氨液,正丁醇液蒸干,残渣加水5 mL使溶解,放冷,通过D101大孔吸附树脂柱(内径1.5 cm,长12 cm),以水50 mL洗脱,弃去水液,再用40%乙醇30 mL洗脱,弃去洗液,继用70%乙醇80 mL洗脱,收集洗液,蒸干,用甲醇溶解并转移至10 mL量瓶中,加甲醇至刻度,摇匀,用0.45 μm的微孔滤膜滤过,即得。
方法(b)供试品制备:与方法(a)同至“正丁醇液蒸干”,用甲醇溶解并转移至10 mL量瓶中,加甲醇至刻度,摇匀,用0.45 μm的微孔滤膜滤过,即得。
方法(c)供试品制备:取本品粉末约4 g,精密称定,置锥形瓶中,加甲醇60 mL,冷浸过夜,加热回流1.5 h,静置5 min,收集上清液,残渣加甲醇40 mL,超声5 min,静置5 min,收集上清液,重复以上步骤3次,合并上清液,回收溶剂并浓缩至干,从“残渣加水10 mL”开始,与方法(a)相同。
方法(d)供试品制备:取本品粉末约4 g,精密称定,置锥形瓶中,加甲醇60 mL,冷浸过夜,加热回流1.5 h,静置5 min,收集上清液,残渣加甲醇40 mL,超声5 min,静置5 min,收集上清液,重复以上步骤3次,合并上清液,回收溶剂并浓缩至干,残渣加热水10 mL,超声溶解,转移至分液漏斗,用水饱和正丁醇提4次,每次50 mL,合并正丁醇液,氨试液洗2次,每次50 mL,弃去氨液,正丁醇液蒸干,残渣用甲醇溶解并转移至10 mL量瓶中,加甲醇至刻度,用0.45 μm的微孔滤膜过滤。
2.3 线性关系的考察 分别精密吸取0.53 mg/mL黄芪甲苷对照品溶液1、3、9、15、21、24 μL注入Waters 2 695,按照上述色谱条件进行实验。以进样质量对数为横坐标,峰面积对数为纵坐标,获得回归方程Y=1.4848974 X+5.062 740,r=0.999 4,表明黄芪甲苷在0.53~12.72 μg时进样量对数与峰面积对数呈良好线性关系。
2.4 稳定性试验 取本品制备供试品溶液,分别于0、2、4、6、8、10、12 h进样10 μL注入Waters 2 695,测定黄芪甲苷峰面积,结果RSD为2.17%,表明供试品溶液在12 h内基本稳定。
2.5 精密度试验 精密吸取0.53 mg/mL黄芪甲苷的对照品溶液10 μL,重复进样6次,测定黄芪甲苷的峰面积,计算,结果其RSD为0.87%。
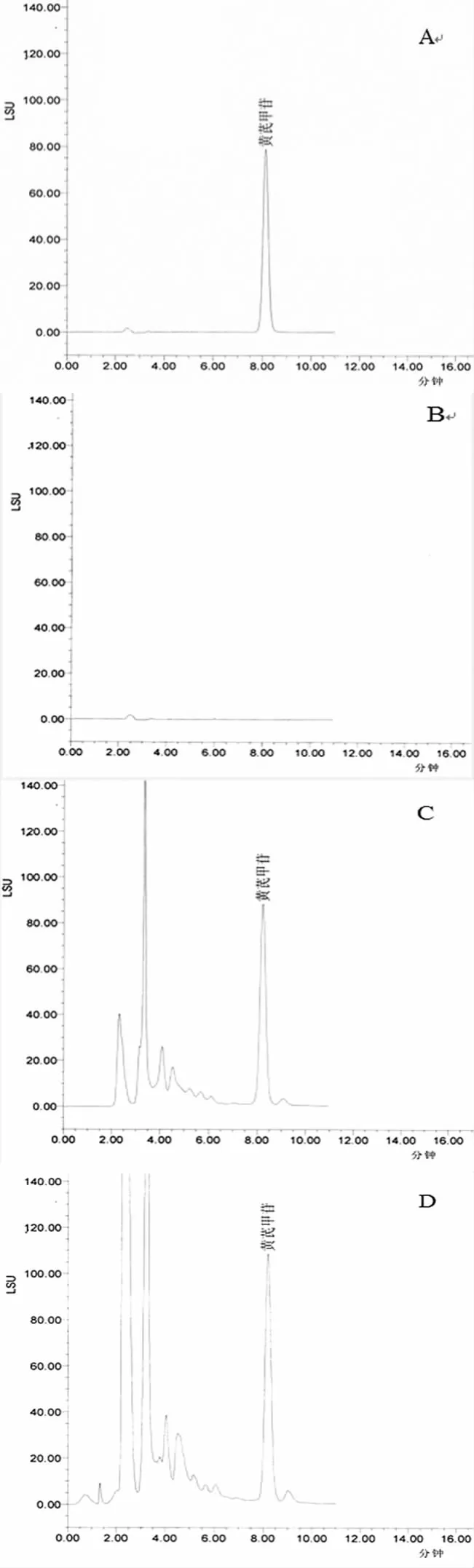
图1 黄芪HPLC-ELSD色谱图:A黄芪甲苷对照品;B空白;C 2010版药典方法;D改进方法(d)
2.6 加样回收率试验 精密称取已知含量的样品5份(批号:20110614,1.052 mg·g-1),每份约4 g,分别精密加入0.53 mg/mL黄芪甲苷对照品2 mL,按供试品溶液制备方法(d)及测定法操作,进行色谱分析,得平均回收率为98.91%,RSD为2.10%。结果见表1。
2.7 样品测定 取同一批号黄芪饮片,按“2.2”项下的方法制备供试品溶液,精密吸取供试品溶液各20 μL注入液相色谱仪,计算样品中黄芪甲苷含量,结果见表2。
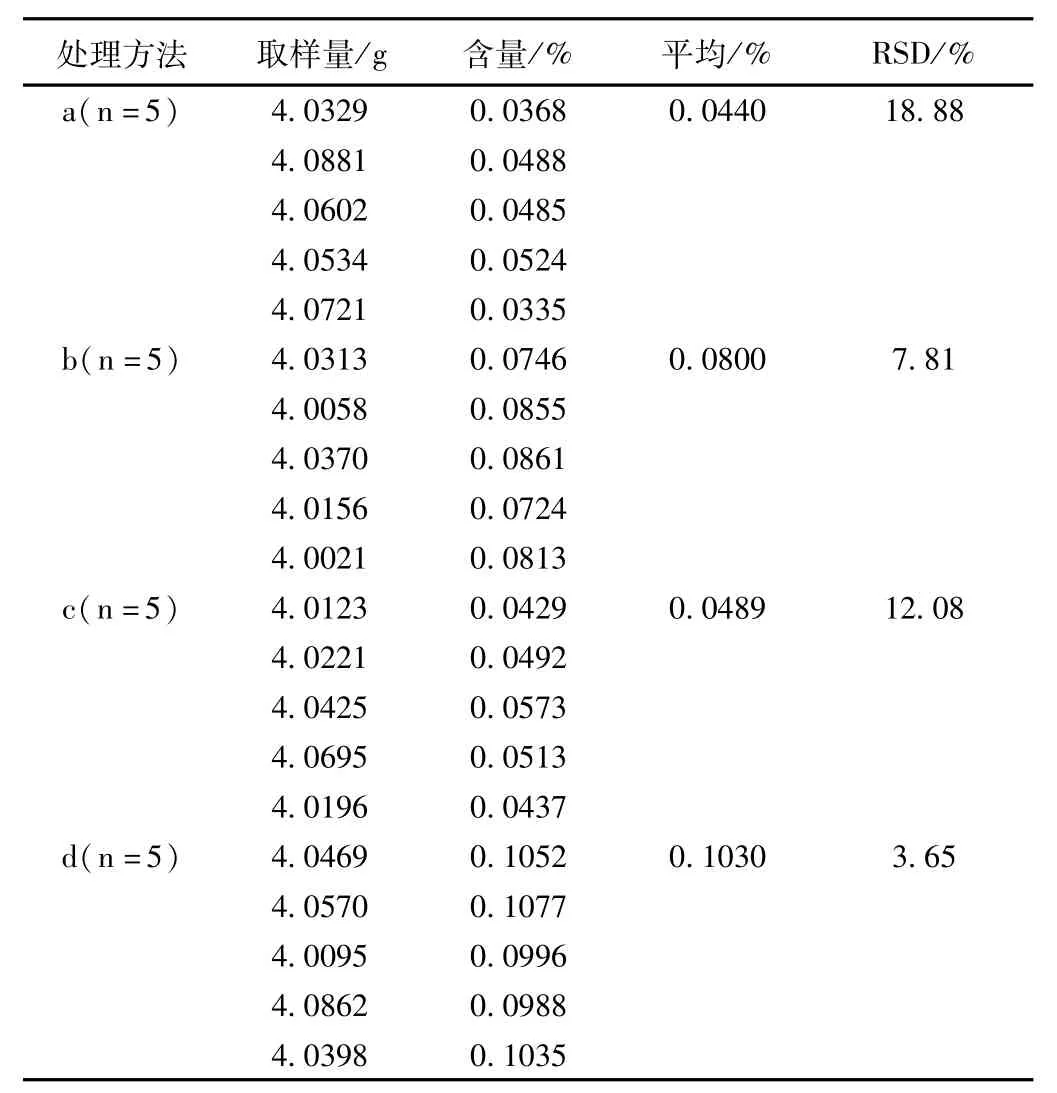
表2 样品中黄芪甲苷的含量测定结果
3 讨论
1)潘细贵[7]已证明,在黄芪供试品制备中D101型大孔树脂吸附除杂步骤即使低浓度的乙醇(15%)也能洗脱出一定量的黄芪总皂苷,并随着浓度的增大而增加,同时D101型大孔吸附树脂对黄芪总皂苷有一定的吸附量。2010版《中国药典》大孔树脂吸附除杂步骤是用40%乙醇30 mL洗脱,所以此方法将洗脱出较多的黄芪总皂苷。实验表明,省去大孔树脂吸附除杂这一步,对黄芪甲苷峰的分离及检测没有任何影响,同时可大幅提高所测样品浓度,提高了测定的准确性、稳定性和重现性。2)结果表明,索氏提取法与加热回流提取法有一定差异,回流提取法提取量稍高。3)样品中黄芪甲苷的含量测定结果的RSD是:方法a>c>b>d。从其结果可以看出现行的2010版《中国药典》由于处理方法的繁琐、步骤多以及其他一些因素的影响,含量测定结果的稳定性、重现性较差。因此我们建议改用具有更好稳定性、重现性的方法(d)。4)《中国药典》要求黄芪甲苷含量0.040%。本实验采用的改进方法(d)所测含量高于标准一倍多,而按《中国药典》方法测定,稍高于标准要求。笔者认为,应该改进实验方法,提高含量标准。5)同《中国药典》采用的方法相比,改进后的方法,样品提取操作简单、方便省时、环保、节约能源与试剂,符合药品检测所要求的简单、方便、高效、准确、重现性好的科学检测理念。可作为黄芪药材、饮片中黄芪甲苷质量的检测方法。6)许多投料黄芪的中成药[8-15],在标准制定黄芪甲苷含量检测时都按照黄芪药材和饮片的方法来处理。所以本实验的改进方法也可为许多中成药中黄芪甲苷质量的检测提供理论依据。
[1]朱燕辉,严丰祥.黄芪甲苷及其生物学活性[J].现代生物医学进展,2008,8(4):781-783.
[2]国家药典委员会.中华人民共和国药典:一部[S].2010版.北京:中国医药科技出版社,2010:283-284.
[3]田南卉,杨国红,方颖,等.高效液相色谱法蒸发光散射检测器测定黄芪和制剂中黄芪甲苷的含量[J].药物分析杂志,2000,20(3):199 -201.
[4]周春玲,鲁静.高效液相色谱一蒸发光散射检测测定黄芪中黄芪甲苷的含量[J].中国中药杂志,2000,25(3):166-168.
[5]纪松岗,李翔,朱东亮,等.高效液相色谱一蒸发光散射检测黄芪药材中黄芪甲苷的含量[J].药学实践杂志,2005,23(5):295-296.
[6]陈有根,辛敏通,杨滨,等.黄芪药材中黄芪甲苷含量测定方法的改进[J].中国新药杂志,2008,17(21):1857-1859.
[7]潘细贵,汪洋,雷湘,等.大孔吸附树脂纯化黄茂总皂昔的提取工艺研究[J].中国医院药学杂志,2005,25(11):1029-1030.
[8]杨瑞瑞,王英,刘艳玲.HPLC-ELSD法测定上林糖泰胶囊中黄芪甲苷的含量[J].安徽医药,2009,13(10):195-196.
[9]谢守兰,李炜,冯敏,等.HPLC-ELSD法测定宫血停颗粒中黄芪甲苷的含量[J].西北药学杂志,2008,23(3):148-149.
[10]涂兴明,李赐恩,吴康郁.高效液相色谱蒸发光检测法测定伤骨健胶囊中黄芪甲苷的含量[J].中药材,2010,33(6):999-1000.
[11]徐新军,胡海燕,王珊.高效液相色谱蒸发光检测器测定新血宝胶囊中黄芪甲苷含量[J].药物分析杂志,2009;29(2):292-294.
[12]杨海燕,徐长根,张倩,等.HPLC-ELSD测定痛宁片中黄芪甲苷的含量[J].中成药,2007,29(3):467-467.
[13]俞建平,方翠芳,唐登峰.高效液相色谱-蒸发光散射法测定肾康宁片中黄芪甲苷的含量[J].中国现代应用药学,2007,24(5):408 -410.
[14]黄丽丹.阿胶养血膏质量标准研究[J].安徽医药,2006,10(11):812-813.
[15]徐忠坤,徐海娟,宋娟,等.HPLC法测定芪葛颗粒中黄芪甲苷[J].中草药,2011,42(1):94-95.
(2012-11-26收稿)
Improved Method for Determination of Astragaloside in Astragali Radix
Ding Ruxian,Li Xia,Li Yijun,Sun Xuemei,Yang Minghua
(Shanghai Pudong Institute for Food and Drug Control,Shanghai 201203,China)
Objective:To establish a simple and reproducible method for determination of astragaloside in Astragali Radix.Methods:Four extraction methods were used and compared.The content of astragaloside was determined by HPLC-ELSD with a Capcell PAK C18(4.6 mm×250 mm,5 um)and a mobile phase of acetonitrile-water(32:68).Results:Using the simple extraction method,the linear relation was good with the range of 0.53~12.72 ug(r=0.999 4).The average recovery was 98.91%with RSD=2.10%.Conclusion:The improved method is simple,accurate,and reproducible and can be used for the determination of astragaloside in Astragali Radix.
Astragali Radix;Astragaloside;Determination;HPLC-ELSD
10.3969/j.issn.1673-7202.2013.06.028
丁如贤,Tel:021-51320025,E-mail:ruxianding@hotmail.com
