PPARγ基因RNA干扰慢病毒载体构建及干扰Leydig细胞株建立
2013-04-13马明月张磊张玉敏裴秀丛段志文
马明月,张磊,张玉敏,裴秀丛,段志文
(1.沈阳医学院公共卫生学院毒理学教研室,辽宁 沈阳 110034;2.大连市疾病预防控制中心)
过氧化物酶体增殖因子激活受体(peroxisome proliferator activated receptors,PPARγ)是核受体超家族成员,在糖类代谢、脂肪酸氧化以及脂肪细胞分化方面有着重要作用[1]。同时,在细胞分化、发育中有重要作用[2-3]。生殖器官中由特定的配体激活后,PPARγ的调节功能通过调节类固醇合成酶的表达能力。各种内源性因素,如花生酸、脂肪酸和前列腺素代谢产物,可作为PPARγ的天然配体。有些环境内分泌干扰物,如邻苯二甲酸二(2-乙基)己酯(DEHP)及其活性中间产物MEHP作为外源性过氧化物酶体增殖剂,在其致生殖毒性中的作用已引起关注[4-5]。
RNA干扰(RNA interference,RNAi)技术是目前最有效的基因功能研究手段之一。慢病毒siRNA表达载体的成功构建使RNAi 技术迈上了新的台阶[6]。研究发现在类固醇合成过程中,睾丸Leydig细胞具有激素分泌功能,且其生成机制均与PPARγ有关[7],故本研究选择小鼠Leydig细胞株TM3,构建特异靶向小鼠PPARγ基因的RNAi慢病毒载体,建立使用PPARγ基因稳定沉默的Leydig细胞株,旨在为研究PPARγ在生殖毒性研究中的功能提供新的实验模型。
1 材料与方法
1.1 细胞培养 小鼠睾丸间质细胞TM3、慢病毒包装细胞,人胚肾细胞293T细胞(购自中国科学院上海生命科学研究院细胞资源中心),常规培养使用含10% FBS (Gibco)的DMEM/F12培养基、DMEM培养基(Gibco)(含1.5 mM L-Glutamine,100 U/ml penicillin,100 μg/ml Streptomycin)中,37 ℃ 5% CO2饱和湿度培养箱中培养。
1.2 针对PPARγ基因的慢病毒载体构建和慢病毒包装 针对Genebank中小鼠PPARγ基因设计的干扰序列,见表1。实验所用慢病毒包装由生工生物工程(上海)公司提供服务。

表1 siRNA序列
寡核苷酸的设计及表达shRNA的慢病毒穿梭质粒的构建:LV3-shRNA模板中的loop结构选用了TTCAAGAGA以避免形成终止信号。正义链模板的5′端添加了GATCC,与BamH I酶切后形成的粘端互补;反义链模板的5′端添加了AATTC,与EcoR I酶切后形成的粘端互补。
根据确定的PPARγ基因RNAi 有效序列sh-PPARγ及对照序列sh-Control,化学合成对应的寡核苷酸DNA单链,退火形成粘性末端的DNA双链,运用基因克隆技术,与经双酶切后的穿梭质粒连接。
1.3 慢病毒质粒共转染293T细胞
1.3.1 慢病毒滴度检测 293T细胞在6 cm细胞培养中培养至80%~90%融合时,倾去培养液,用3 ml D-Hank′s液洗涤细胞2次。加入1 ml Trypsin-EDTA 液,混匀后,小心弃去胰酶溶液,37 ℃放置3~5 min。再加入2 ml 含10% FBS的DMEM/F12培养液,吹打使细胞形成单细胞悬液。将细胞稀释至3×105个/ml,按3×104个/孔的浓度接种96孔板,混匀后于37 ℃ 5% CO2培养24 h。将慢病毒原液(10~20 μl),用10% FBS的DMEM/F12培养液10倍稀释3~5个梯度(根据细胞状态,如有必要可加入终浓度为5 μg/ml的 Polybrene)。吸去96孔板中的培养液,每孔加入100 μl稀释的病毒液,同时设立空白对照组,于37 ℃ 5% CO2培养24 h。吸弃96孔板中的稀释病毒液,每孔加入150 μl 10% FBS的DMEM/F12培养液(根据细胞状态,如有必要可分出1/3~1/5)于37 ℃ 5% CO2继续培养48、72 h。通过荧光显微镜计数荧光细胞,结合稀释倍数计算病毒滴度(实验过程中注意生物安全要求)。
1.3.2 靶细胞转染实验 TM3细胞在10 cm 细胞培养皿中培养至80%~90%融合时,倾去培养液,用2 ml D-Hank’s液洗涤细胞2次。加入1 ml Trypsin-EDTA液,混匀后,37 ℃放置2~3 min。小心弃去胰酶溶液,再加入2 ml 含10% FBS的DMEM/F12培养液,吹打使细胞形成单细胞悬液。血球计数板计数,按1×105个/孔的浓度接种96孔板,混匀后于37 ℃ 5% CO2培养24 h。将慢病毒原液200 μl,用10% FBS的DMEM/F12培养液5倍稀释(根据细胞状态及类型,如有必要可加入终浓度为5 μg/ml的 Polybrene)。吸去96孔板中的培养液,每孔加入上述100 μl稀释的病毒液,同时设立空白对照组,于37 ℃ 5% CO2培养24 h。吸弃96孔板中的稀释病毒液,每孔加入200 μl 10% FBS的DMEM/F12培液(根据细胞状态,如有必要可分出1/3~1/5) 于37 ℃ 5% CO2继续培养72、96 h,分别收样,所得细胞用于mRNA及Western blot检测。
1.4 Real-time PCR 检测 病毒转导后,制备实验组、对照组、未处理组细胞cDNA,使用QIAGEN公司RNA提取试剂盒提取转染筛选后的细胞RNA及逆转录反应合成cDNA。取上述总RNA 1 μg进行逆转录,具体方法同我们之前的研究[8],引物对序列由本室设计,生工生物工程(上海)公司合成(表2)。根据Real-time PCR相对定量2-△△CT方法,计算目的基因的相对表达,重复3次。
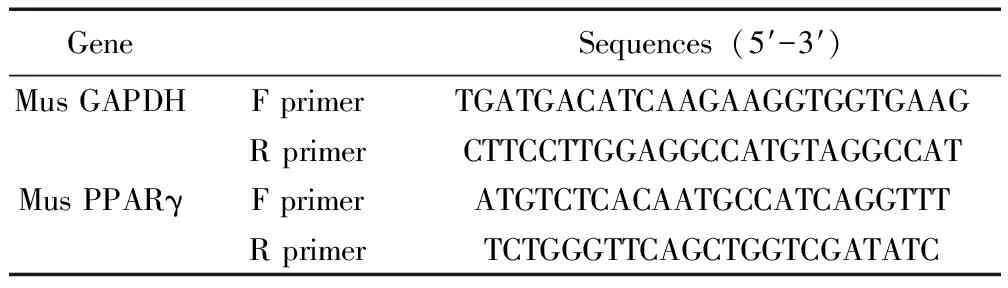
表2 引物序列
1.5 Western blot 检测 转导病毒后,收集各组细胞,每个细胞样本加入100 μl 细胞裂解液,使组织完全裂解。裂解物移入离心管中。取5 μl 样本加入5 μl 2×SDS-PAGE上样缓冲液(loading buffer),100 ℃加热处理5 min,冰上冷却。12 000 g 离心10 min去除不溶性沉淀。样品使用10% SDS-PAGE分离,每孔上样量为20 μl。电泳结束后,将PVDF膜在甲醇中浸泡1 min,再使用转移缓冲液(transfer buffer)浸泡凝胶、滤纸和在甲醇中浸泡过的PVDF膜10 min,然后制备转移三明治。使用Semi-Dry Cell 进行半干电泳转移,转移条件为30 mA 70 min。用1×Ponseau 液染色并标记蛋白质Marker的条带位置。使用Blocking Buffer 封闭转印膜2 h,然后用1×TBST 洗涤3 次,每次10 min。加入适当稀释的一抗,4 ℃温育过夜。用1×TBST 洗涤3 次,每次15 min。加入适当稀释的二抗,室温温育2 h。用1×TBST 洗涤3 次,每次15 min。用Super Signal West Pico Chemiluminent Substrates进行化学发光检测,并对X光片曝光。经显影定影处理后,胶片用凝胶成像分析系统拍照,分析图像,比较各组间PPARγ蛋白的相对表达量。重复3次。
2 结果
2.1 特异靶向小鼠PPARγ基因的RNA干扰慢病毒载体LV3-sh-PPARγ构建成功 测序结果,如图1。直接测序结果显示表明,重建的穿梭质里包含sh-PPARγ序列,说明成功构建包含PPARγ-sh-RNA的新的慢病毒载体,命名为LV3-sh-PPARγ。由图1可见,靶序列1087、1089、1091构建成功;靶序列1092碱基丰度不够,且出现错误序列。
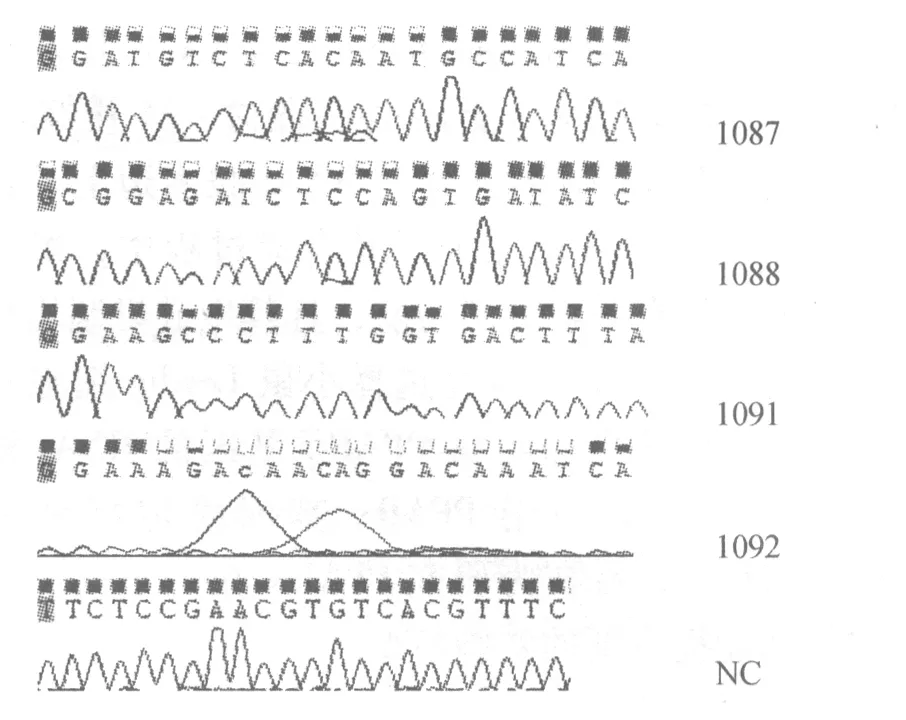
图1 靶序列(1087、1088、1091、1092)及对照序列(NC)的测序结果
2.2 病毒包装产生有效滴度的病毒液转导TM3细胞转导效率高 慢病毒浓缩成1 ml后,取10 μl按照1∶10侵染293T细胞,以检测包装病毒滴度情况。转染72 h后,用荧光显微镜观测293T细胞的GFP,以估计侵染效率,四种靶序列及对照序列的慢病毒载体共转染293T细胞效率高。滴度合适即可进行下步转导试验。经计算,病毒的滴度为5×108TU/ml。转染72 h后,用荧光显微镜观测细胞的GFP,具有RNAi效应的细胞(1087、1088、1091、1092)转染效率>90%。
2.3 LV3-sh-PPARγ能有效降低PPARγ基因在Leydig细胞TM3中的表达 转导后,Real-Time PCR相对定量结果,见图2。实验组(1087、1088、1091、1092)TM3细胞PPARγ的mRNA的表达比未处理细胞下降70%(P<0.05),对照组与未处理细胞差异无显著性(P>0.05)。Western blot 显示实验组(1087、1088、1091、1092)PPARγ蛋白表达量比未处理细胞明显下降(P<0.05),对照组与未处理细胞差异无显著性(P>0.05)。LV3-sh-PPARγ对于内参GAPDH无影响。见图3、4。
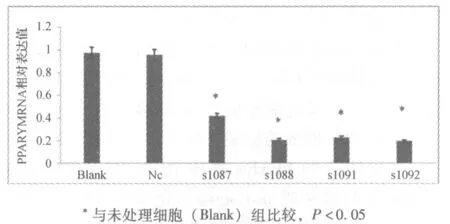
*与未处理细胞(Blank)组比较,P<0.05图2 转导72 h后 PPARγ mRNA相对表达水平

1:样本1087;2:样本1088;3:样本1091;4:样本1092;5:NC;6:Blank图3 TM3细胞中PPARγ的蛋白表达水平(GAPDH作为内参照)
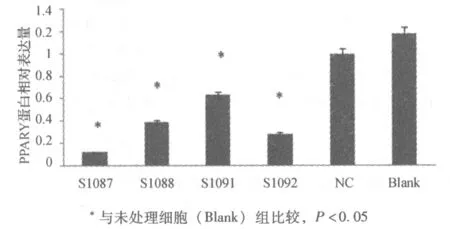
*与未处理细胞(Blank)组比较,P<0.05图4 TM3细胞中PPARγ蛋白相对表达量(GAPDH作为内参照)
2.4 PPARγ稳定沉默的小鼠Leydig细胞株TM3构建成功 转导慢病毒后的TM3细胞荧光阳性率达到90%以上,通过分拣阳性转导细胞,得到稳定转导LV3-sh-PPARγ以及LV3-Control两个细胞株,分拣后的细胞荧光阳性率达100%。继续培养分拣后细胞,当细胞生长至最佳状态后,收集并冻存细胞,保存于-150 ℃。这样PPARγ基因稳定敲低的Leydig细胞株TM3及其对照细胞株TM3构建成功。
3 讨论
构建稳定干扰PPARγ基因的细胞株是人们关注的焦点,目前对于PPARγ在睾丸间质细胞中扮演的角色仍然存在着很大的争议。构建稳定干扰PPARγ基因的细胞株,进而揭示PPARγ在睾丸间质细胞中的确切功能,不仅能帮助阐明睾酮分泌机制,并且能开拓基因治疗的一条崭新途径,PPARγ将可能成为极好的药物潜在治疗靶点[9]。慢病毒介导RNAi载体的研究与其他基因功能研究技术相比,RNAi具有明显优势。RNAi的抑制效率远高于反义核苷酸及核酶技术,并且作用更稳定,采用表达载体可使作用更持久[10]。本研究构建了四个靶序列从基因测序结果、mRNA表达及蛋白表达检测结果来看,靶序列1088的效果较好。本研究第1次采用慢病毒介导RNAi技术构建特异靶向PPARγ基因的慢病毒RNA干扰系统,并构建PPARγ稳定敲低的人睾丸间质细胞株,为该基因功能的研究成功地构建了有效的细胞模型,也为相关的基因治疗开辟了崭新的前景。
参考文献:
[1]Semple RK,Chatterjee VK,ORahilly S.PPAR gamma and human metabolic disease [J].J Clin Invest,2006,116(3):581-589.
[2]Komar CM.Peroxisome proliferator-activated receptors(PPARs) and ovarian function:implications for regulating steroidogenesis,differentiation,and tissue remodeling [J].Reprod Biol Endocrinol,2005,3:41-55.
[3]Kersten S,Desvergne B,Wahli W.Roles of PPARs in health and disease [J].Nature,2000,405(6785):421-424.
[4]Lovekamp ST,Davis BJ.Mechanisms of phthalate ester toxicity in female reproductive system[J].Environ Health Perspect,2003,111(2):139-145.
[5]马明月.PPARs介导邻苯二甲酸酯雌性生殖毒性的研究进展[J].工业卫生与职业病,2009,35(5):310-312.
[6]Nishitsuji H,Ikeda T,Miyoshi H,et al.Expression of small hairpin RNA by lent virus -based vector confers efficient and stable gene suppression of HIV-1 on human cells including primary non-dividing cells[J].Microbes Infect,2004,6(1) :76-85.
[7]Kowalewski MP,Dyson MT,Manna PR,et al.Involvement of peroxisome proliferator-activated receptor γ in gonadal steroidogenesis and steroidogenic acute regulatory protein expression[J].Reprod Fertil Dev,2009,21(7):909-922.
[8]马明月,张玉敏,裴秀丛,等.青春前期邻苯二甲酸二乙基己酯(DEHP)暴露对雌性大鼠生殖发育及过氧化物酶体增殖剂激活受体(PPARs)的影响[J].卫生研究,2011,40(6):688-697.
[9]郑素平,洪晓婷,王琴,等.以siRNA 表达载体稳定抑制缝隙连接蛋白43表达的睾丸间质细胞和睾丸支持细胞系的建立[J].Chinese Pharmacological Bulletin,2010,26(10):1285-2009.
[10]Scherer L,Rossi J.Recent applications of RNA interference (RNAi) in mammalian systems[J].Letters in Peptide Science,2003,10(3-4):255-267.
