Raney Ni催化合成3-甲基吲哚的方法研究
2013-03-31茹婷婷
茹婷婷
(吉林建筑工程学院基础科学部,长春 130118)
吲哚是一种重要的精细化工原料,广泛应用于医药、农药、香料、染料、食品和饲料添加剂等领域[1].尤其3-甲基吲哚具有更广泛的应用,其具有强烈的粪臭味,但稀释后具有优美的花香味,常用于茉莉、柠檬、紫丁香、兰花和荷花等人造花精油的调合剂[2].3-甲基吲哚也是重要衍生物和化学合成中间体,吲哚衍生物,如吲哚乙酸、吲哚-3-丁酸是重要的广谱性植物生长调节剂,目前主要用于促进草本和木本观赏植物插枝的生根[3].许多生理活性很强的天然物质均为吲哚的衍生物,如中成药六神丸中的蟾酥就含有5-羟基吲哚衍生物,许多生物碱中也含有吲哚环系,常用降压药物利血平就是吲哚的重要衍生物[4-5].所以,3-甲基吲哚的简便、经济合成方法的研究,一直是研究工作者关注的热点.吲哚传统的制备方法是采用煤焦油分馏法,由于资源有限,而且分离装置繁杂、能耗大,因而化学合成技术应运而生.本文提出了一种操作简便,产量高,耗能低,污染小的合成3-甲基吲哚的方法.
1 实验部分
1.1 试剂和实验仪器
邻硝基乙苯(AR);二甲亚砜(CP);多聚甲醛(AR);液体石蜡;甲醇钠(AR);Raney Ni VARIAN UNITY-500 MHz核磁共振仪(内标TMS,溶剂CDCl3);MAGNA—IR 560型红外光谱仪(KBr压片法);Agilient 1100 LCMsD型质谱仪;GSA-0.25型250毫升高压反应釜;PE-2400自动元素分析仪;电天平;旋片式真空泵;三用紫外线分析仪;旋转蒸发仪;79-l型磁力加热搅拌器;高效薄层板;柱层析分离柱.
1.2 3-甲基吲哚的合成路线

1.2.1 2-(2-硝基苯基)-丙-1-醇的合成
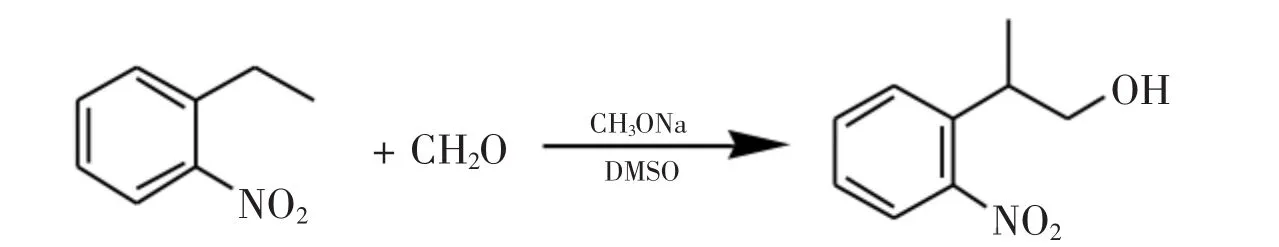

图1 2-(2-硝基苯基)-丙-1-醇的红外谱图
将15.1 g(0.1mol)邻硝基乙苯,20 mL二甲亚砜,1.5 g(0.05 mol)多聚甲醛,依次放入100 mL三口烧瓶中.在搅拌下,升温至50℃,期间不断加入甲醇钠甲醇饱和溶液以维持pH为9(共加入5 g).反应45 min后,停止加热,向反应体系中加入浓盐酸调pH至中性.后处理:液相分析(254 nm)结果表明,2-(2-硝基苯基)-丙-1-醇峰面积百分比31.85%,其他为原料邻硝基乙苯(几乎无副反应产物).减压蒸馏所得的反应液得到棕红色2-(2-硝基苯基)-丙-1-醇纯品.邻硝基乙苯共计回收10.1 g,邻硝基苯乙醇共计得到5.7 g(按已转化的邻硝基甲苯计重量收率为114%,摩尔收率为95.1%).IR(KBr):3 547,3 374,3 073,2 974,2 937,2 878,1 608,1 576,1 523,1 355,1 195,1 037,1 013,853,785,748,711 cm-1;13C NMR(500 MHz,CDCl3)δ:150.87,138.39,132.92,128.47,127.37,124.29,67.88,36.59,17.78;1H NMR(500 MHz,CDCl3)δ: 7.767~7.737(dd,J=1.2,8.1 Hz,1 H,ArH),7.611~7.556(dt,J=1.5,4.5 Hz,1 H,ArH),7.512~7.481 (dd,J=1.5,8.1 Hz,1 H,ArH),7.392~7.335(m,1 H,ArH),3.788(br s,2 H,CH2),3.550~3.482(m,1 H,CH),1.725(br s,1 H,OH),3.171~3.139(d,J=6.9 Hz,3 H,CH3).图1为2(2-硝基苯基)-丙-醇的红外谱图.
1.2.2 2-(1-羟基丙-2-基)苯胺的合成
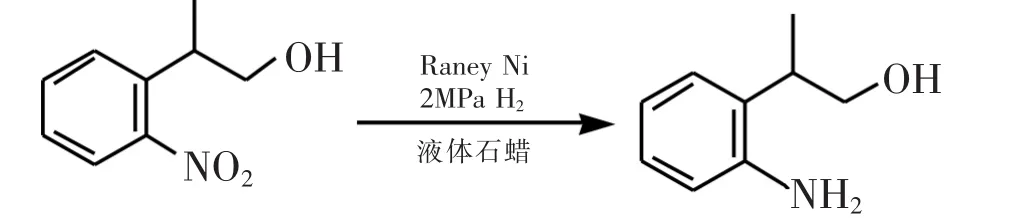

图2 2-(1-羟基丙-2-基)苯胺的红外谱图
将20 g 2-(2-硝基苯基)-丙-1-醇,100 mL液体石蜡加入至GSA-0.25型250 mL高压反应釜,加入2.12 g Raney Ni催化剂.试漏,用4×2 MPa氢气赶气.开始搅拌,升温至90℃,通氢压至2 MPa反应.4.5 h后,基本上不吸氢,反应结束.卸压,卸出反应液,过滤除去Raney Ni催化剂后静置,分层,分液,无水硫酸镁脱水后得到棕红色2-(1-羟基丙-2-基)苯胺15.28 g,收率91.6%,HPLC纯度>99%.IR(KBr):3 378,3 065,3 031,2 965,2 929,2 874,1 624,1 583,1 497,1 453,1 381, 1 298,1 255,1 155,1 094,1 034,1 314,751 cm-1;13C NMR(500 MHz,CDCl3)δ:144.86,129.41,127.37,126. 61,119.74,116.84,68.67,35.70,16.84;1HNMR(500 MHz,CDCl3)δ:7.137~7.083(dt,J=1.2,8.1 Hz,1 H,ArH),7.062~7.032(dd,J=1.5,7.5 Hz,1 H,ArH),6.847~6.794(dt,J=1.2,7.2 Hz,1 H,ArH),6.731~6. 701(dd,J=1.5,7.8 Hz,1 H,ArH),3.814~3.652(m,4 H,CH2and NH2),3.147~3.078(m,1 H,CH),1.591 (br s,1 H,OH),1.283~1.259(d,J=7.2 Hz,3 H,CH3).图2为2-(1-羟基丙-2-基)苯胺的红外谱图. 1.2.3 3-甲基吲哚的合成

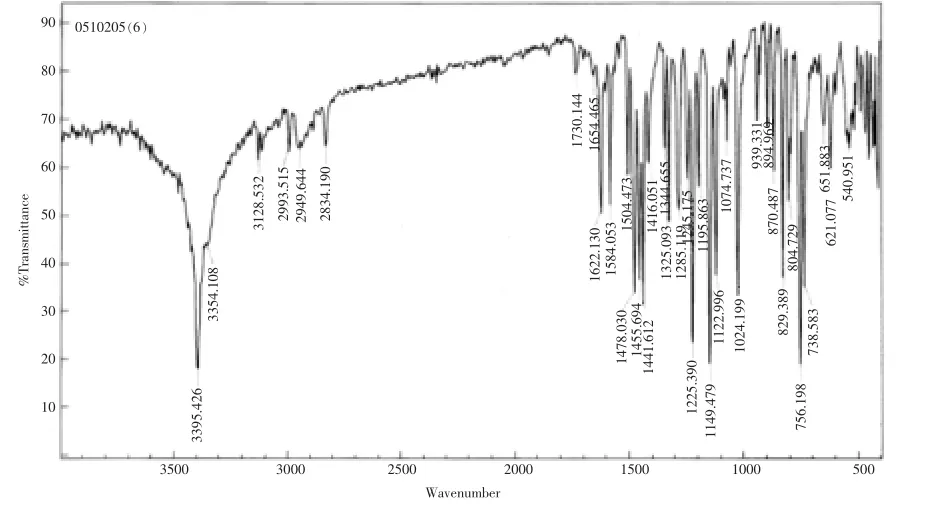
图3 3-甲基吲哚的红外谱图
取10.39 g 2-(1-羟基丙-2-基)苯胺,1.5 g Raney Ni催化剂,10 mL液体石蜡放于50 mL三口瓶中.三口瓶装备有接液器,球形冷凝管,机械搅拌和温度计.氮气置换整个体系后,开始搅拌,升温至220℃反应,TLC监控反应进程;4.5 h后反应完成,共收集馏出液共计1.5 g.将体系冷却至80℃,趁热过滤掉Raney Ni,HPLC分析反应液转化率100%,选择性92.4%.滤液冷却后从液体石蜡中析出黄色鳞片状晶体(3-甲基吲哚粗品),重7.2 g,依此计算的收率79.9%.柱色谱分离(乙酸乙酯:石油醚=5%~30%梯度洗脱),可得到白色晶状固体3-甲基吲哚.IR(KBr):3 408,3 053,2 925,2 858,1 454,1 345,1 300,1 246,1 229,1 084,1 006,798,739 611,580 cm-1;13C NMR(500 MHz,CDCl3)δ:136.47,128.50,122.09,121.78,119.34,119.07,111.98,111.16,9. 91;1HNMR(500 MHz,CDCl3)δ:7.861(s,1 H,NH),7.598~7.570(dd,J=0.6,7.2 Hz,1 H,ArH),7.360~7. 330(dd,J=1.2,7.8 Hz,1 H,ArH),7.218~7.094(m,2 H),6.971~6.961(dd,J=0.9,1.5 Hz,1 H,ArH),2. 340~2.336(d,J=1.2 Hz,3 H,CH3).图3为3-甲基吲哚的红外谱图.
2 结果与讨论
2.1 空间位阻和电子效应对反应的影响
邻硝基乙苯的苄基位连有一个甲基,甲基的供电效应使苄基位氢更不容易被碱夺除;另外,相邻甲基的空间位阻同样使羟甲基化反应变得困难.这两种情况的存在都使苄基位反应活性不高,可能需要更强的碱才能催化该反应.所以在简单而不成功地尝试了20%NaOH水溶液(收率仅4.1%)后,采用醇钠催化羟甲基化反应获得了成功(2-(2-硝基苯基)-丙-1-醇单程收率提高至31.85%).由于苄基上已经有一个甲基,再加上一个羟甲基后,由于空间位阻较大,很难在同一位置继续发生二缩反应,因此,该反应的选择性比较好.尝试过一次邻硝基乙苯的羟甲基化放大实验(邻硝基乙苯投料量1 100 g),但由于条件没控制好(可能是因为二甲亚砜含水,将醇钠催化剂分解的原因),2-(2-硝基苯基)-丙-1-醇单程收率只有18%左右.即便如此,通过该反应也获得了足够量的2-(2-硝基苯基)-丙-1-醇纯品用于下步反应.这样,我们采用氯霉素工业大量副产物邻硝基乙苯为原料,经过羟甲基化、催化氢化,以及催化环合三步反应,成功地合成了3-甲基吲哚,总收率69.6%.这为邻硝基乙苯的利用开辟了一条新的途径,具有较高的环保和经济价值.
2.2 反应机理的研究
对于邻氨基苯乙醇在Raney Ni催化剂下生成吲哚的机理这一问题,瑞士联邦理工学院的Hammerschmidt认为,邻氨基苯乙醇环合机理是先脱水环合成吲哚啉,然后再脱氢成吲哚.而日本的Watanabe等也研究了邻氨基苯乙醇的环合反应,他们认为,反应是先脱氢,再环化为吲哚.笔者认为,这一反应的机理应属于上述两种机理之一,至于具体是哪一种,还有待实验论证.
本文对这一问题进行了验证,Hammerschmidt所提出机理涉及的中间体吲哚啉可以很方便从市场上买到,便于验证,所以,本文先假设反应是按照Hammerschmidt所提出的先脱水,再环合的机理进行.首先采用市购的吲哚啉测定了其在Raney Ni催化剂下的最低脱氢温度,实验条件为10.0 g吲哚啉,2.0 g Raney Ni,50 mL液体石蜡.用氮气置换体系后,开始搅拌并加热,从50℃开始,每隔10℃左右稳定一段时间(15 min左右),取样分析,以确定吲哚出现的最低反应温度.实验结果表明,在上述反应条件下,吲哚啉在90℃~100℃左右开始脱氢生成吲哚.随后本文又测定了邻氨基苯乙醇生成吲哚的最低反应温度.实验条件和操作方法同吲哚啉脱氢实验.实验结果表明,在上述反应条件下,邻氨基苯乙醇在110℃左右开始生成吲哚.这两个实验说明,吲哚啉的脱氢温度和邻氨基苯乙醇生成吲哚的温度(如果是按照Hammerschmidt的机理进行,可以认为是脱水温度)相近,如果反应是按照Hammerschmidt所提出的先脱水,再环合的机理进行的话,那么,在体系中应该有可能检测出吲哚啉的存在.但在该实验条件下,并没有发现体系中有吲哚啉的存在.由此看来,其机理很可能与Watanabe所提出的一样,即先脱氢,再环合成吲哚.
根据所得到的实验结果,我认为在Raney Ni催化下,邻氨基苯乙醇的环化机理如图4所示.

图4 Raney Ni催化下邻氨基苯乙醇环化机理
3 结语
此合成路线借鉴了Watanabe吲哚[6-7]合成及Ryuichi[8]的邻氨基苯乙醇环合条件,以邻硝基乙苯为原料,首先用甲醛对苯环上的乙基进行羟甲基化合成邻硝基苯乙醇,然后采用Raney Ni为催化剂催化邻硝基苯乙醇氢化还原,以及邻氨基苯乙醇的闭环,最终合成了3-甲基吲哚,三步反应总收率77.7%.并且对反应空间位阻和电子效应进行了讨论,为邻硝基乙苯的利用开辟了一条新的途径,也提供了一种操作简便,产量高,耗能低,污染小的合成3-甲基吲哚的方法.
[1]吴立军,吴继洲,王锋鹏.天然药物化学[M].北京:人民卫生出版社,2003:367-370.
[2]Saxton J E.Indoles[M].New York:John Wiley,1983,876-882.
[3]Yoshio B,刘 湘.吲哚生物碱药物[J].国外医药,1989,4(2):54-68.
[4]杨秀伟.天然产物化学手册-生物碱[M].北京:化学工业出版社,2005:228-312.
[5]Prabhakar C,Kumar N V,Reddy M R,et al.Process Research and Development of Melatonin[J].Organic Process Research&Development,1999,3(2):155-160.
[6]Richou R M,Lallouette P,Richou H.Anti-inflammatory activity of inflammatory ubstances.Action of abrin and saponin on inflammation induced with taphylococcal toxin[J].C.R.Acad.Sci.,1967,264(20):2426-2428.
[7]Chakeaborty A,Chowdhury B K,Bhattacharya P.Clausenol and Clausenine-twocarbazole alkaloids from Clausena anisata[J].Phytochemistry,1995,40(7):295-298.
[8]Harada T,Katsuhira T,Hattori K,et al.Stereoselective synthesis of gem-Disubstituted cyclopropanes from gem-dibromocyclopropanes[J].Tetrahedron Lett.,1989,159(30):6035-6038.
