难熔元素钨在NiAl位错体系中的占位及对键合性质的影响*
2013-02-25陈丽群于彭小芳
陈丽群于 涛 彭小芳 刘 健
1)((中南林业科技大学理学院,长沙 410004)
2)((钢铁研究总院功能材料研究所,北京 100081)
(2012年10月22日收到;2013年2月4日收到修改稿)
1 引言
NiAl金属间化合物具有较高的熔点(1638°C)、较低的密度(5.90 g/cm3)、较高的杨氏模量 (240 GPa)、较高的热导性 (70—80 W/m·K),以及优良的抗高温氧化性等特点.且晶体结构为体心立方基超点阵,结构简单,对称性高,进行塑性变形的潜力大,因此可以预期用作理想的高温结构材料.但是,NiAl合金的低温塑性差和高温强度低这两个致命的缺点制约了它的实际应用.因此,材料科学工作者通过很多技术去拓展它的性能,如合金化、制备多相合金、制备复合材料、定向凝固、机械合金化、热压及热等静压、燃烧合成以及微晶涂层等工艺,大大改善了NiAl合金的力学性能[1],其中合金化是用来改善NiAl合金力学性能的一种非常有效的方法之一[2-4].通过合金化来改变这种金属间化合物的几何和电子结构、键合性质以及长程有序化的程度等来改善其室温塑性和增强高温强度[5,6].
Tang等[4]利用XRD,SEM,EDX等方法研究了Ti,Hf,Nb及W对NiAl-Cr(Mo)金属间化合物的微观结构和力学性能的影响,结果发现添加元素Ti,Hf,Nb和W对合金的高温屈服强度和室温屈服强度及韧性有大的改善.Djajaputra等[7]对NiAl中轻杂质 (B,C,N,O,Si,P,S)杂化进行了第一性原理研究,发现N,O使材料脆化,而B,C与近邻基体Ni和Al原子间形成了共价键使得NiAl金属间化合物的结合加强,Si,P,S的成键性质与B,C类似.近年来有关NiAl合金化的研究较多[8,9],但主要集中在完整晶体中的合金化,而材料的性能受多种因素的影响,如杂质含量、结构缺陷等.杨等[10-13]研究发现,NiAl合金拉伸延展性的增强与区域位错的形成及运动密切相关.因此了解合金元素与位错之间的相互作用对改善材料性能具有重要的意义,位错与杂质元素之间的相互作用机理有两种成熟的观点:其一是对于大多数合金,掺杂原子和基体原子大小失配是主要的因素;另一种观点则认为掺杂原子的电子结构起主要的作用,因为NiAl合金的机械性能主要依靠增加的微量合金.首先一个合金原子会改变基体原子的电子结构和电子间的相互作用等特征;反过来,也会影响能级特征和能级的跃迁.
Kontsevoi等[14]使用real-space紧束缚线性muf fin-tin轨道的方法研究了在NiAl中〈100〉(010)刃型位错和掺杂元素(Ti,V)之间的相互作用,计算结果表明:在位错芯区基体原子电子态被发现主要通过化学相互作用和电子相互作用两种机理引起杂质位错相互作用.这个结果很好地解释了为什么NiAl合金的固溶强化效应同杂质的电子结构有关而不是与大小失配有关.最近陈等[15,16]用第一性原理方法研究了NiAl中轻杂质原子(B,C,N)与位错的相互作用,结果表明:轻杂质原子同它近邻的基体原子有强烈的相互作用,杂质原子对位错有较强的钉扎效应,这个结果被期待可以提高NiAl合金的强度.近年来,虽然NiAl合金的研究备受关注,但有关从电子层次对位错和杂质之间相互作用的研究报道很少.因难熔元素最重要的优点是有良好的高温强度,对熔融碱金属和蒸汽有良好的耐蚀性能.同时Wang等[17]研究发现NiAl合金中绝大多数的错位都是Burgers矢量为b=〈100〉的刃型位错,因此本文对难熔元素W与〈100〉(010)刃型位错之间的相互作用进行第一性原理研究,第一性原理研究方法广泛地应用于金属[18]、合金[19]及金属间化合物[20]的电子结构计算.
2 模型与方法
使用分子动力学方法获得NiAl中〈100〉(010)刃型位错平衡构型[15,16].其结构具有C2v的对称性,在垂直位错线方向,〈100〉(010)刃型位错有两个不等价的(001)原子面(分别称作A和B),在位错线方向的堆垛顺序是ABAB,位错的原子构型如图1所示.很明显,位错芯区的原子构型不同于完整晶体,位错的出现使材料结构发生改变,很可能会导致材料的性质发生大的变化.
由于A原子面的原子既可以是Ni原子也可以是Al原子,因此可建立二个原子模型,当A原子面上的原子是Ni原子时,称该模型为Ni心位错芯,当A原子面上的原子是Al原子时,称该模型为Al心位错芯.为了研究难熔元素W对〈100〉(010)刃型位错电子结构的影响,将W引入位错芯.基于使杂质原子有尽量高的局域对称性,且只考虑单杂质模型,因此将W替换图1中标注为1的原子.电子结构计算模型沿位错线方向取七个原子面,包括杂质原子共有209个原子.采用DMol[21,22]方法对掺杂模型进行结构优化,在优化过程中,只对图1中标注阿拉伯数字的原子及等价原子进行弛豫.能量梯度和原子位移的收敛判据分别为0.001 Ry/a.u和0.001,结合能收敛到0.00001 Ry,如果各原子轨道集居数在相邻两步自洽迭代中差值的方均根收敛至10-5,则表示结果收敛.

图1 〈100〉(010)刃型位错的平衡原子构型,实心圆和空心圆符号分别表示堆垛原子面A(·),B(°)上的原子
本文采用离散变分方法(DVM)[23]计算未掺杂位错(纯位错)体系和掺杂位错体系的电子结构,该方法是在密度泛函理论框架下,求解Kohn-Sham方程:

该方法成功地应用于金属[24]、合金[25]及金属间化合物[26]的电子结构计算.
3 结果与讨论
3.1 能量分析
杂质偏聚能(Eseg)可以用来描述杂质对体系稳定性的影响,Eseg定义为[27]
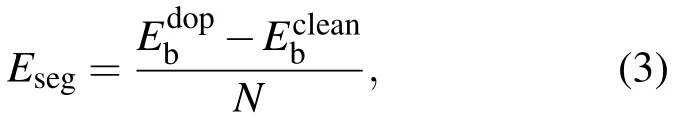

表1 NiAl位错体系的结合能Eb和杂质偏聚能Eseg
由表1可知,与不掺杂体系相比较,掺杂体系的结合能更低,这反映了掺杂元素有利于体系的稳定.由杂质偏聚能可以看出,Al位错芯的Eseg比Ni位错芯的Eseg低得多,几乎相差一半,这表明掺杂元素W优先占据Al位错芯中的Al格位,这一结果与Song等人[28]的研究结果一致,他们用离散变分方法研究了加入有序化合物NiAl中的合金化元素对其化合物电子结构及性能的影响,通过计算NiAl-X(X=Ti,V,Cr,Zr,Nb,Mo,Hf,Ta,W,Si,Ga)合金体系的电子结构及结合能发现:所有元素有强的Al格位择优占位优势,因此下面只计算Al位错芯体系的电子结构.
3.2 原子间相互作用能
在固体物理中,常用结合能表征原子的结合强度,但这只具有统计意义,它不能反映在微观尺度上结合力与晶体方向的依赖关系.基于量子力学Green函数理论,Wang发展建立了原子间相互作用能的数学表述[29],即用计算给出的群表象下的本征矢矩阵及相应的哈密顿矩阵,通过表象变换建立原子轨道表象下的波矢矩阵及哈密顿矩阵,从而给出原子间相互作用能.原子l和m间的相互作用能为

原子间相互作用能类似于键序(Bond Order),但包含了哈密顿相互作用矩阵,所以它既反映了原子间电子云的交叠,同时也反映了原子间的轨道杂化,Elm非常具体地反映了原子间的相互作用的情况.通常情况下,两原子间相互作用能的数值负得越多,表明两原子间相互作用越强.
掺W位错体系中位错芯中一些原子对的相互作用能呈现在表2中,为了比较,纯位错芯体系中对应原子对的相互作用能也同时在表2中给出.为了更清楚地看出难熔元素W对NiAl合金性能的影响,掺杂体系与纯位错体系之间的相互作用能的变化也给在表2中.

表2 纯位错芯和掺杂位错芯中被选原子对的原子间相互作用能(单位eV),其中Eclean和Eimp分别表示纯位错芯和掺杂位错芯中被选原子对的原子间相互作用能,ΔEW=Eimp-Eclean.1号原子在纯位错芯中表示Al原子,而在掺杂位错芯表示杂质原子W.阿拉伯数字对应于图1中的原子序号
从表2可以看出除了Ni10-Ni11原子对之外,掺杂原子和它近邻原子之间的相互作用能明显增加,这就意味着与纯位错体系相比较,用W替换Al原子可以提高原子间的相互作用能.另外,在掺杂体系中基体原子之间的相互作用也增加了(如Al3-Al4和Ni6-Ni9原子对).尤其是掺杂原子与其最近的基体原子(Ni6,Ni10)和次近邻的基体Al3原子的相互作用比在纯错位体系中1号原子(Al)和与之相对应的Ni6,Ni10和Al3原子间的相互作用强很多.因此,在掺杂体系中杂质原子可以大大地提高原子间的相互作用,使得掺杂位错体系的运动比纯位错体系更困难,这可能有利于NiAl合金强度的提高.而掺杂体系中Ni10-Ni11原子对的相互作用能比纯为位错体系仅少了0.02 eV,近似没变,这可能是因为这对原子离杂质原子W较远,这也说明了杂质原子影响的局域性.
3.3 态密度
由于是在实空间中求解Kohn-Sham方程,得到的本征能级是分立的,而实际固体中能级应为准连续的.为了模拟这种连续性以获得态密度(DOS)[30],DVM中用Lorentz展宽方法,定义原子n,l轨道的分波态密度(PDOS)为

态密度与原子间的成键特性密切相关,利用DVM计算得到的PDOS如图2所示,图2(a)和(b)分别为纯位错体系和掺W位错体系中芯区原子的PDOS.比较图2(a)和(b)发现有二个明显的特点,一是两体系中位于反键区最大多数是p轨道和较少的s轨道,而d轨道几乎都位于成键区,这与最外层价电子的性质一致;二是掺杂体系中每个原子的PDOS曲线较纯位错体系对应原子的PDOS曲线都朝更低能级移动,特别是Al3原子3s3p轨道和Ni6、Ni10原子的3d4s轨道移动的幅度更大,这意味着掺杂体系中位错芯区原子的价电子移到更深能级,并与杂质原子W的价电子参与了成键,使得杂质原子与近邻基体原子间的相互作用加强.此外,从图2(a)看到,对于纯位错体系中心原子Al1的3s3p轨道与近邻Al3原子的3s3p及Ni6和Ni10的4s轨道有强的杂化,而与Ni6和Ni10的3d4p轨道几乎没有相互作用.对于掺杂体系,从图2(b)可看出,难熔元素W替换Al1原子后,主要是W原子的4d轨道与近邻Al3原子的3p轨道及Ni6、Ni10原子的3d轨道杂化,而与Al3原子的3s轨道及Ni6、Ni10的4s4p轨道几乎无相互作用.由此可见,W替换Al1后,主要由于杂质原子W的4d轨道与近邻基体原子Al3的3p轨道及Ni6、Ni10的3d轨道杂化,使得位错芯区难熔原子与近邻基体原子间相互作用加强,
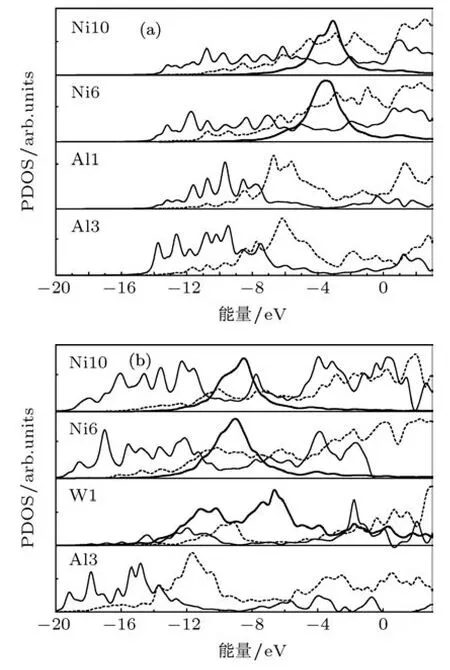
图2 杂质原子和其近邻原子的PDOS曲线 (a)纯位错体系;(b)掺W位错体系(厚实线和薄实线及虚线分别表示d,s和p轨道,Fermi能级平移到零)
3.4 差分电荷密度
为了更直观地了解难熔元素W对NiAl合金性能的影响,计算了掺杂位错芯体系的差分电荷密度分布,其定义为

从图3(a)可以看出,在掺杂位错芯体系中可以看到明显的电荷重新分配.电荷分布主要集中在杂质原子 W 和邻近原子 (Al3,Al4,Al8,Al12)之间,这表明在掺杂原子和它邻近的Al原子之间形成了很强的化学键.另一个特点是掺杂原子W和Al原子之间的相互作用集中在杂质原子附近的小区域内.

图3 掺杂位错芯体系包含杂质原子W的(001)原子面和(100)原子面的差分电荷密度分布 (a)(001)原子面;(b)(100)原子面.等高线间隔为0.002 e/a.u3,实线和虚线分别表示得到电荷和失去电荷
从图3(b)也可以看到,掺杂原子W与它近邻的基体原子之间有电荷的积累.这意味着沿着位错线方向掺杂原子和其近邻基体原子(Ni)间的相互作用也加强了,电荷密度分析的结果与原子间相互作用能的结果是一致的.以上对原子间相互作用能、态密度和电荷密度的分析都反映出,难熔原子对NiAl中〈100〉(010)位错芯有较强的钉扎效应,这必然也会影响镍铝合金的力学性能.
4 结论
本文利用 DMol方法计算了 NiAl〈100〉(010)二种几何位错芯模型的结合能和杂质偏聚能,二个掺杂体系的结合能较纯位错体系的结合能都低,且Al位错芯体系的杂质偏聚能比Ni位错芯低2.21 eV,这表明难熔元素W的添加使体系更加稳定,且W倾向于替代Al位错芯体系中的Al原子.利用DVM方法仅研究了难溶元素W对NiAl〈100〉(010)Al位错芯电子结构的影响,计算了原子间相互作用能、态密度及电荷密度分布.原子间相互作用能,计算结果表明:与纯位错体系比较,在掺杂体系中杂质原子与基体原子间相互作用增强了,尤其是掺杂原子与其最近邻的基体原子间(Ni6,Ni10)和次近邻Al3原子间的相互作用明显强于在纯位错体系中Al1原子和与其对应的基体原子间的相互作用;同时掺杂体系中,基体原子本身之间的相互作用也有所加强.态密度和电荷分布的结果表明:在W原子与其邻近的基体原子之间有较多的电荷聚集,这说明W原子与近邻基体原子形成了较强的化学键,且原子间相互作用的加强,主要是由于W原子的4d轨道与近邻基体Al原子的3p轨道及Ni原子的3d轨道之间的杂化.W原子与错位相互作用的加强可能有利于材料强度的提高.总之.难熔元素W的加入,大大改变了NiAl合金的能量及电子结构,从而影响位错的运动及材料的性能.
[1]GuoJ T,Ren W L,Zhou J 2002 Acta Metall.Sin.38 667(in Chinese)[郭建亭,任维丽,周健2002金属学报38 667]
[2]Darolia R 1991 JOM 43 44
[3]George E P,Liu C T 1990 J.Mater.Res.5 745
[4]Tang L Z,Zhang Z G,LiS S,Gong S K 2010 Trans.Nonferrous Met.Soc.China 20 212
[5]Shang J X,Yu X Y 2008 Acta Phys.Sin.57 2380(in Chinese)[尚家香,喻显扬2008物理学报57 2380]
[6]Shang J X,Yu T B 2009 Acta Phys.Sin.58 1179(in Chinese)[尚家香,于潭波2009物理学报58 1179]
[7]Djajaputra D,Cooper B R 2002 Phys.Rev.B 66 205108
[8]TakasugiT,KishinoJ,Hanada S 1993 Acta MetAll.Mater.41 1009
[9]Hu X L,Zhang Y,Lu G H,Wang T 2009 J Phys.Condens Matter.21 025402
[10]Pollock T M,Lu D C,ShiX,Eow K 2001 Mater.Sci.Eng.A 317 241
[11]Yang C 2006 Int.J.Mech.Sci.48 950
[12]Ghosh B,Crimp M A 1997 Mater.Sci.Eng.A 239-240 142
[13]EbrahimiF,Shrivastava S 1998 Acta Mater.46 1493
[14]KontsevoiO Yu,Gornostyrev Yu N,Freeman A J,Katsnelson M I.,Tre filov A V 2001 Philos.Mag.Lett.81 455
[15]Chen L Q,Yu T 2011 Sci.China Physics,Mechanics&Astronomy 54 815
[16]Chen L Q,Qiu Z C 2011 Defect and Diffusion Forum 318 23
[17]Wang Y L,Jones I P,Smallman R E 2006 Intermetallics 14 800
[18]Fan X J,Liu F Q,Yin D 2005 Chin.Phys.14 2287
[19]Feng H J,Liu F M 2008 Chin.Phys.Lett.25 671
[20]LiQ X,LiZ Y,Zhu Q S,Yang J L,Hou J G 2001 Acta Phys.Sin.50 1877(in Chinese)[李群祥,李震宇,朱清时,杨金龙,侯建国2001物理学报50 1877]
[21]Delley B 1991 J.Chem.Phys.94 7245
[22]Delley B 1990 J.Chem.Phys.92 508
[23]Guenzburger D,Ellis D E 1992 Phys.Rev.B 45 285
[24]Chen L Q,Wang C Y,Yu T 2006 J.Appl.Phys.100 023715
[25]Wang S Y,Wang C Y,Sun J H,Duan W H,ZhaoD L 2001 Phys.Rev.B 65 035101
[26]Dang H L,Wang C Y,Yu T 2007 J.Appl.Phys.101 083702
[27]Yan J A,Wang C Y,Duan W H,Wang S Y 2004 Phys.Rev.B 69 214110
[28]Song Y,GuoZ X,Yang R,LiD 2001 Acta Mater.49 1647
[29]Wang C Y 1995 Defect Diffus.Forum 125-126 79
[30]Delley B,Ellis D E,Freeman A J,Bearends E J,Pos D 1983 Phys.Rev.B 27 2132
