三羰基茂卤钼复合物的结构及Cp…Mo作用的研究
2013-01-19邝代治冯泳兰张复兴许志锋庾江喜蒋伍玖曾兰玲
邝代治,冯泳兰,张复兴,许志锋,庾江喜,蒋伍玖,曾兰玲
(1.衡阳师范学院 化学与材料科学系,湖南 衡阳 421008;2.功能金属有机材料湖南省普通高校重点实验室,湖南 衡阳 421008)
环戊二烯负离子与羰基钼构成半夹心状的钼负离子[1],钼负离子在卤代反应时,既有亲核性又可有亲电性[2]。在环戊二烯基羰基钼金属有机化学中,[Mo(C5H5)(CO)3]-负离子是一类重要的中间体,通过研究其电子结构、空间构型和成键特征,了解具有η5配位的三羰基茂环卤钼复合物及其电子效应等,阐明其化学性能具有重要意义。
近年来,通过实验方法对η5,6配位的金属有机配合物的研究成绩显著,然而,关于这类金属配合物的理论研究滞后于实验,许多新型配合物主要由实验方法获得。随着研究的深入,一些实验结果需要理论解释,以进一步指导实验。近年来国内外化学工作者注意到了这一问题,已有较多报道[3],主要集中于茂、苯环配位体系的研究。η5,6配合物具有丰富的结构和性能,且中心金属和配位不同,其配合物的性能差异较大,因此,深入研究该类配合物的结构,在理论上对其化合物的谱学性质和化学反应性进行解释,有助于指导实验进一步合成新配合物、开发功能新材料,并为有机化学、结构化学、合成化学、生物化学等作出贡献。
量子化学方法,已成为理论上探讨物质分子结构与性质的重要工具[4]。近些年来,随着计算机的快速发展,对于许多分子体系,在分子的几何结构、电子性质、能量及光谱性质等方面计算出与实验非常吻合结果,可以较好地预测一些结构和化学反应性能等。因此,量子化学计算方法使人们从理论上探讨物质的结构和性质已成为可能。本文用量子化学方法研究标题复合物(C5H5)Mo(CO)3[(X:F(Ⅰ),Cl(Ⅱ),Br(Ⅲ),I(Ⅳ)]的电子结构和成键特征等,以期阐明其结构与反应性能的关系。
1 研究方法
通过GaussView对(C5H5)Mo(CO)3(X=F,Cl,Br,I)分子模型,如图1所示(图中的原子标记为计算的编号)。采用HARTREE-FOCK方法在LANL2DZ基组水平上对结构进行全优化,振动频率分析结果未出现虚频,构型体系稳定。计算涉及18原子,139原子基函(氟代复合物140),357个初始函数(氟代复合物370),其中有49个占据分子轨道(氟代复合物50)。然后对优化体系计算单点能,获取分子轨道的组成、单元结构的能量、原子电荷和Wiberg键级等。全部计算使用Gaussian03w[5]程序包,在P4微机上完成。
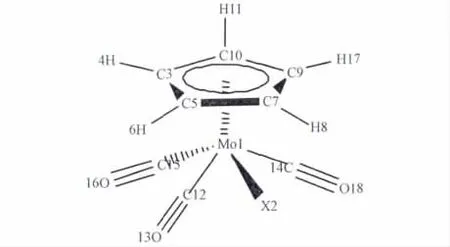
图1 (C5H5)Mo(CO)3X分子模型Fig.1 Molecular structure of(C5H5)Mo(CO)3complex
2 结果与讨论
2.1 复合物的体系能和稳定性
振动分析和优化计算结果表明,优化结构的振动光谱未见虚频,表明优化结构具有一定的稳定性。复合物的单点体系能和前沿分子轨道能见表1。
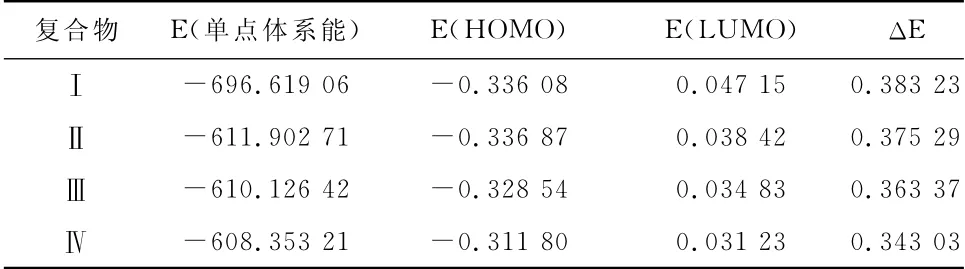
表1 复合物的单点体系能和前沿分子轨道能(a.u.)Table 1 The single point energy and the energy of frontier MO
根据分子轨道理论,前线轨道和相近分子轨道的能量对于复合物的稳定性起着重要作用[6]。对于结构类似的分子,体系总能量和占据分子轨道能量越低,结构单元越稳定,体系总能量和占据轨道的能量均表明复合物基态稳定。但从表1数据显示,它们之间的单点体系能有差异而稳定性不尽相同,其稳定性依次:(Ⅰ)>(Ⅱ)>(Ⅲ)>(Ⅳ)。复合物的前沿分子轨道能E(HOMO)均较低,与前沿最低未占轨道的能量间隙ΔE[E(LUMO)—E(HOMO)]在0.34~0.39a.u.之间,分子的最高占据轨道(HOMO)能级和最低空轨道(LUMO)能级之差ΔE也反映出复合物的稳定性,且HOMO能级越低,差值ΔE越大,从HOMO上电离一个电子越困难,从氧化还原的角度,复合物难以失去HOMO的电子,尤其是氟代复合物难以失去HOMO的电子,相对碘代复合物较容易失去HOMO的电子。
比较它们的E(HOMO)、E(LUMO)和ΔE发现,ΔE(Ⅰ)>ΔE(Ⅱ)>ΔE(Ⅲ)>ΔE(Ⅳ),其中碘取代复合物ΔE值较小,说明此复合物激发电子相对另三个卤代物容易,即碘取代复合物相对稳定性最差,这与碘的原子半径较大、碘化物容易发生氧化还原有关。图2分别为三羰基(X)钼复合物的前沿分子轨道示意图(X分别为F、Cl、Br和I)]

图2 三羰基(卤)钼复合物复合物的前沿分子轨道示意图Fig.2 The schematic diagram of frontier MO fo(C5H5)Mo(CO)3
2.2 构型和键级
表2、3为优化所得复合物的部分键参数。结果表明,优化所得的构型中,金属原子位于茂环(Cp)的上方,Mo与Cp上碳原子的“键长”均在0.237nm~0.249nm之间,属于相互共价作用。金属钼原子与茂环中心(Cp*)的“键长”随Ⅰ(0.213 07nm)、Ⅱ(0.212 62nm)、Ⅲ(0.212 53nm)、Ⅳ(0.212 18nm)变小,即Mo…Cp*的作用距离依次变小,茂环(Cp)与钼的配位作用依次加强。在茂环(Cp)与钼的配位作用加强的同时,复合物中Mo-X键长随X(Ⅰ、Ⅱ、Ⅲ、Ⅳ)依次增长,即Mo-X键长:R(Mo-I,0.298 276 8nm)>R(Mo-Br,0.279 170 8nm)>R(Mo-Cl,0.262 073 2nm)>R(Mo-F,0.208 286 6nm),显然,Mo-I键最长,碘-钼复合物相对稳定性最差,而Mo-F键最短,F-Mo复合物的相对稳定性最好。中心钼原子与羰基的作用也呈规律变化,Mo-CO键长随Ⅰ、Ⅱ、Ⅲ、Ⅳ而减小,羰基对钼的配位作用随Ⅰ、Ⅱ、Ⅲ、Ⅳ增强。复合物(Ⅰ)、(Ⅱ)、(Ⅲ)、(Ⅳ)中,环碳原子共轭面良好,二面角D在±0~1.2°之间,也表明Mo与Cp的作用较强,成为稳定复合物,Mo-Cp*为Mo金属到茂环平面中心的垂直距离。
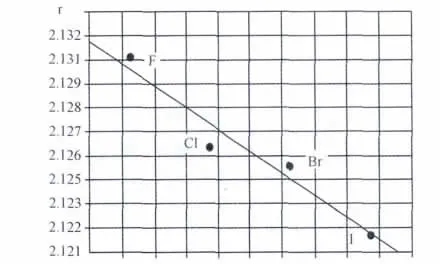
图3 Mo…Cp*的作用距离(r×10nm)Fig.3 The bond lengths of the Mo…Cp*
计算所得金属原子(Mo)与配体原子之间的Wiberg键级列于表2.从表可见茂环碳C3、C5、C7、C9和C10与中心Mo原子的键级相近,并随氟、氯、溴和碘代复合物的键级增大,键级于0.21~0.30之间,具有较多的共价性质,虽然Mo-C较弱,但5个Mo-C键之和形成强作用,因此,成为稳定的η5复合物;羰基碳C12、C14和C15与中心Mo原子的键级在0.65~0.75,表明Mo-CO具有较强的结合作用,并随氟、氯、溴和碘代复合物的键级增大,羰基向钼配合依次加强,与它们之间的键参数一致。
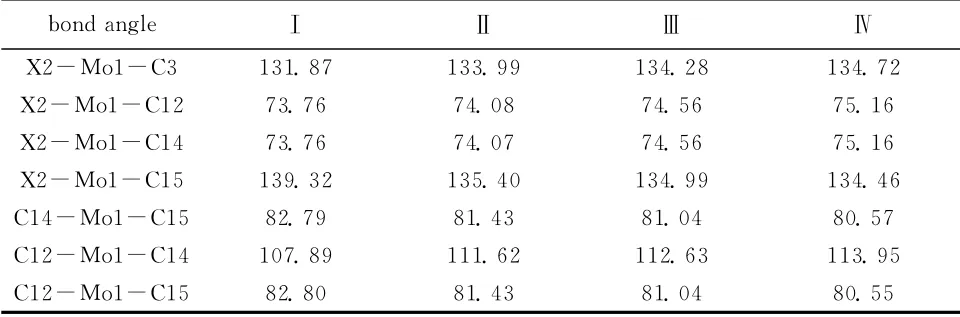
表3 复合物的主要键角(°)Table 3 The geometric parameters of complexe[bond angle(°)]
2.3 复合物电子结构研究
分子中原子的电荷分布是影响化学反应活性的重要因素。在标题复合物中,茂环和羰基分别与钼配位,钼原子还与卤原子成键。改变其中的卤素原子将影响到钼原子上的电荷分布,从而影响茂环和羰基的电荷分布。钼原子上的电荷分布反映其亲电(核)性能。
为探讨卤素原子取代的标题复合物的电荷转移,在同一条件下计算其电子结构,得复合物(Ⅰ)、(Ⅱ)、(Ⅲ)和(Ⅳ)的原子电荷分布,表4可见,①卤素原子、环戊二烯碳、羰基氧均带负电荷,且相对应原子带电子多少分别为:(Ⅰ)>(Ⅱ)>(Ⅲ)>(Ⅳ),C(7)原子却例外,在C(7)的对应复合物的原子电荷由(Ⅳ)>(Ⅲ)>(Ⅱ)>(Ⅰ)。②环戊二烯氢带0.268~0.295左右正电荷,相对应原子失电子多少分别为:(Ⅰ)>(Ⅱ)>(Ⅲ)>(Ⅳ)。③钼原子在(Ⅰ)、(Ⅱ)呈正电,在(Ⅲ)、(Ⅳ)呈负电,Mo原子带电荷从氟到碘代物由正电荷到带负电荷变化,表明Mo原子所带电子逐渐增加,从氟到碘代物中Mo获得附近配体更多的电子。羰基氧原子虽带负电荷,但羰基碳带正电荷,两者所带电荷之和为正电荷,表明整个羰基逐渐失去电子,且直接与Mo原子相连的羰基随氟到碘代物电荷密度依次减少。
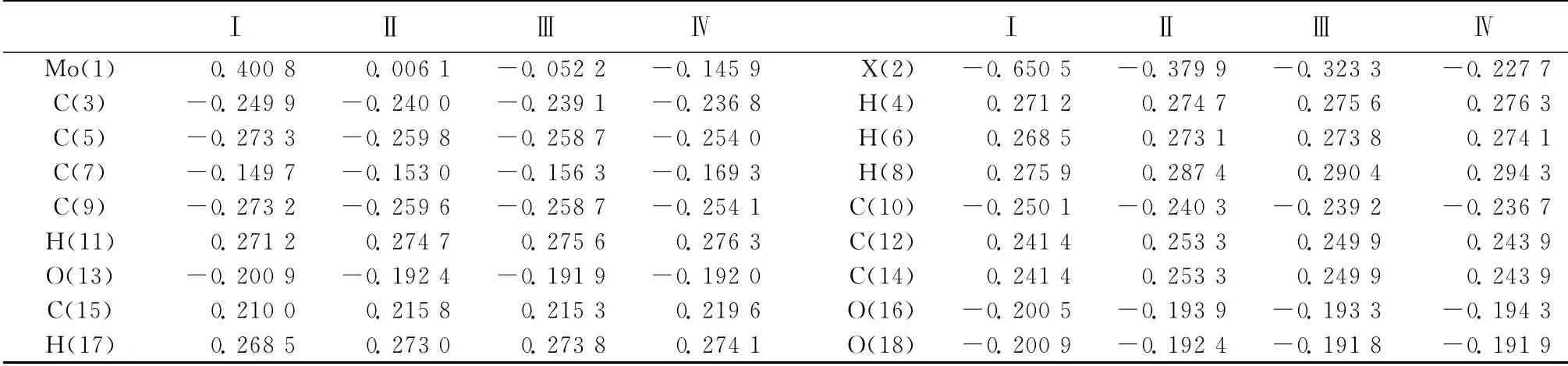
表4 复合物的原子电荷布局Table 4 Atomic charge population of the complexes
环戊二烯负离子带-1e,与钼配合后,环戊二烯的碳、氢原子所带电荷之和均变为带正电,分别为+0.1591,+0.2302,+0.2372和+0.2442,环戊二烯碳上的p电子发生离域向钼原子的d轨道转移,形成电荷转移复合物。且这种电子转移随氟、氯、溴、碘代物增加,因此,表现为钼原子所带电荷由正到负,前两者钼表现为亲电性,后两者钼表现为亲核性,三羰基环戊二烯基卤钼复合物既可有亲电性,又可具亲核性。
2.4 分子轨道组成研究
为了探索标题复合物的结构及成键特征,运用参与组合的各类原子的轨道系数的平方和,归一化后,来表示该类原子在分子轨道中的贡献。把复合物中的原子分为6类:(1)钼原子Mo;(2)卤原子X;(3)茂环碳原子C(I);(4)羰基碳原子C(II);(5)羰基氧原子O;(6)氢原子H。复合物的分子轨道成分如图4所示,它们具有类似的分子轨道组成谱。表5列出了复合物分子前沿分子轨道组成。由表5可见,最高占据轨道中以钼原子(其中Mo原子的d轨道对占据分子轨道的贡献为主,p轨道贡献较少,s轨道的贡献几乎为零)及卤原子占有成分为主,表明钼与卤原子之间的作用较强。其次是茂环碳原子轨道(主要由茂环碳的p轨道)对HOMO的贡献,表明Mo与Cp*作用也较强,因此,钼原子与环茂二烯之间是通过p-π和d-π作用形成类似于“氢键”作用,其HOMO和LUMO轨道的立体图如图2所示。
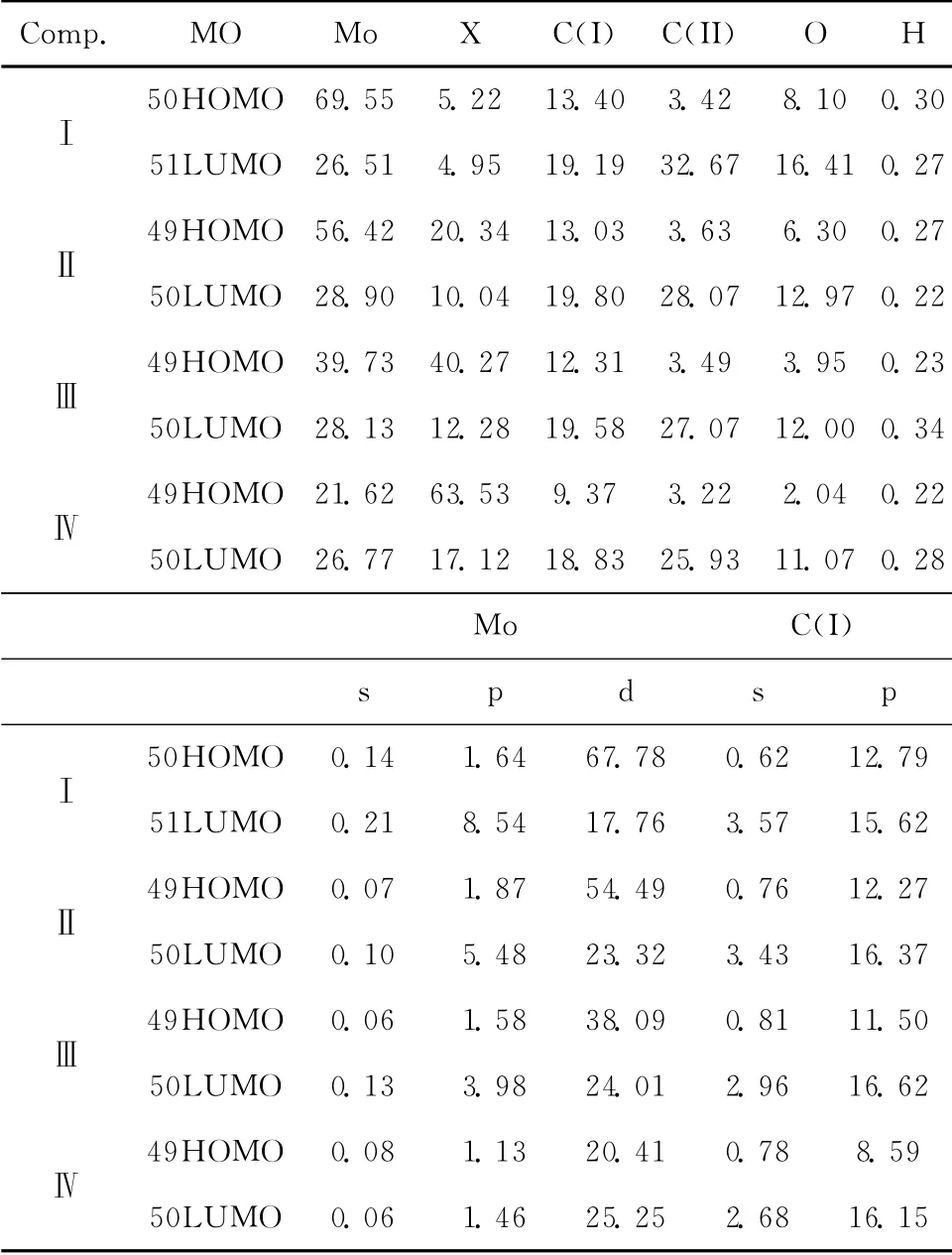
表5 复合物的前沿分子轨道组成(%)Table 5 The composition of the frontier molecular orbitals of the complexes(%)

图4 复合物的分子轨道成分图Fig 4. The composition of the molecular orbitals of the complexes
3 结 论
运用量子化学方法,研究三羰基环戊二烯基卤钼复合物的分子结构、电子结构、以及Cp-Mo的作用情况,茂环、羰基和卤原子与金属钼属强配合作用,形成稳定的复合物,随氟、氯、溴到碘代复物展现出规律变化。较好的阐明茂钼复合物中钼原子与环戊二烯的作用和结构。为羰基茂钼金属复合物的设计和组装提供理论参考。
[1](a).邝代治,周秀中,徐善生.三羰基(五甲二硅基)环戊二烯基 钼 衍 生 物 的 合 成 和 结 构[J].化 学 学 报,1993,51(10):1035
(b).邝代治,周秀中.四羰基二(五甲二硅基环戊二烯基)二钼的合成和反应[J].化学学报,1994,52(9):853
(c).邝代治,周秀中.双(三甲硅基环戊二烯基羰基钼)配合物的合成和结构[J].化学学报,1995,53(3):243
(d).邝代治,周秀中.Study on the reactivty of tricarbonyl(pentamethyldisilanyl)cyclotadienyl molybdenum anion[J].Chinese chemical letters,1993,3(12):1015
(e).邝代治,周秀中.三羰基全甲硅基环戊二烯基钼衍生物的合成与表征[J].应用化学,1995,2,1-12
[2](a).周秀中,邝代治,徐善生.溴钼环卡宾配合物的合成及结构[J].有机化学,1993,13(6):627
(b).邝 代 治,周 秀 中.[(η5-C5H4Si2Me5)(CO)(PPh3)2MoBr]络合物的合成[J].化学通报,1993,5:30
(c).邝代治,周秀中.三羰基(五甲二硅基)环戊二烯基卤钼络合物合成的改进[J].化学通报,1993,9:37
[3](a).Asirvatham,V.S.;Gruhn,N.E.;Lichtenberger,D.L.;Ashby,M.T.Electronic Factors for Protonation of an Organometallic Molecule.Photoelectron Spectroscopy and Electron ParamagneticResonanceStudyof[(η6-C6H6)Mo(TRIPOD)]0/+[J].Organometallics,2000,19:2215-2227.
(b).Valery I.Faustov,Mikhail P.Egorov,Oleg M.Nefedov and Yuri N.Molin.Ab initio G2and DFT calculations on electron affinity of cyclopentadiene,silole,germole and their 2,3,4,5-tetraphenyl substituted analogs:structure,stability and EPR parameters of the radical anions[J].Phys.Chem.Chem.Phys..2000,2(19):4293-4297.
(c).Brisdon A.K.,Crossley I.R.,Pritchard R.G.Tricarbonyl(η6-chlorobenzene)chromium[J].Acta Cryst.2003,59:322-324.
(d).Yeung,L.K.;Kim,J.E.;Chung,Y.K.;Rieger,P.H.;Sweigart,D.A.Control of Ligand Substitution and Addition Reactions of(Arene)Cr(CO)3Complexes by Attachment of a Self-Closing Redox Switch[J].Organometallics,1996,15,3891-3896.
[4]徐光宪,黎乐民,王德民.基本原理和从头计算法[M].北京:科学出版社,1985.
[5]Aeleen F,Michael J F.Gaussian98User's Reference Gaussian,Inc.,Garnegie Office Park,Bldg.6Pittaburgb,PA15106USA.
[6]周公度,段连运.结构化学基础[M].3版.北京:北京大学出版社,2003.
