碳受体配位键解离能的密度泛函理论计算
2013-01-17张秀娟庞先勇
张秀娟,庞先勇
(1. 山西大同大学化学与环境工程学院,山西大同037009;2. 太原理工大学化学化工学院,山西太原030024)
键解离是化学反应的基本步骤,对化学反应起着决定性的影响。 键解离能是化学反应热效应的主要部分,对热效应的精确计算一直是理论化学家追求的目标之一[1],而精确计算键解离能是达到该目标的可靠保证[2]。包括Hartree-Fock 自洽场以及相关能校正方法在内的量子化学理论以往在计算反应热和键解离能方面是成功的[3],这一事实表明量子化学是达到这一目标的得力工具。密度泛函理论是一种研究多电子体系电子结构的量子力学方法。密度泛函理论在物理和化学上都有广泛的应用,特别是用来研究分子和凝聚态的性质,是凝聚态物理和计算化学领域最常用的方法之一[4]。尽管如此,随着研究工作的深入,特别是类似于本文涉及的特殊共价键的发现,对这些数据的精确度要求也越来越高,以至于应用其中不同的方法对它们进行计算,会出现结果不一致的情况[5]。
配位化合物不同于通常的化合物,是通过由一方(配位体)提供电子对,而另一方(中心体)提供空轨道,并接受电子对而成键,称之为配位键的方式结合在一起的[6]。 最新研究发现表明,碳存在一种新型配合物,在该配合物中碳原子不像以往发现的碳作为电子给体的情况,而是作为电子受体的[7]。 对这种新型配位键的解离能计算,对计算方法以及密度泛函理论中不同泛函的选取提出了更高的要求。本文采用目前流行的7 种密度泛函对这类配合物进行构型优化,结合能计算以及C←N,C←P 两种键解离能计算,以比较它们的稳定性。在此基础上,对计算结果与实验值或高精度G4 计算值进行了比较,来评价密度泛函方法的表现。
1 方法
包括配位键的一般共价键的化学键解离是指在气相条件下发生下列反应:

式中,R-X 是反应物,R 和X 是生成的自由基或原子。
而R-X 的键解离能(bond dissociation energy,BDE)D(R-X) 也可以由反应物和生成物的标准生成焓推导出:
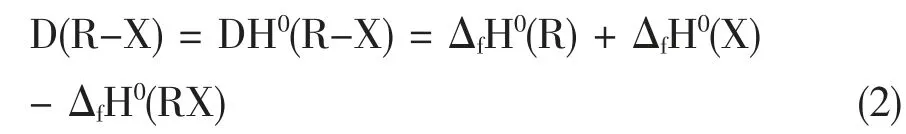
式中,D(R-X)表示化学键解离能,DH0(R-X)中的H强调解离能属于热化学焓变;上标“O”表示在标准条件下;ΔfH0(X),表示X 物种的标准生成焓(1 标准大气压,298.15 K)
对所有分子或自由基的构型,在B3LYP/6 -311 + g(d,p) 水平上进行优化。对优化后构型,在同水平进行频率分析来得到焓、Gibbs 自由能等热力学数据和振动零点能(zero point energy,ZPE)数据,并通过有无虚频确认构型是稳定构型[8]。 零点能校正因子为0.987 7。分子的焓值计算方法为:
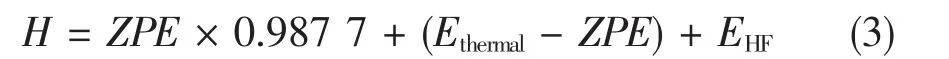
7 种密度泛函方法(DFT)包括B3LYP,B2PLYPD,B2PLYP,B3,BMK,M05-2X,M06-2X,与高精度的G4 方法,都应用Gaussian09 软件包进行计算。
2 结果与讨论
以多种理论计算4 种碳配合物:H3N→C←NH3,(CH3)N→C←N(CH3),H3P→C←PH3,(CH3)P→C←P (CH3) 的键解离能,以实验值和高精度G4 值作为参考,来评价密度泛函方法的表现。
通过比较几种DFT 方法来计算的键解离能大小,来判断键的稳定性。该体系中的碳与传统上所理解的碳不同,传统的碳经激发后看有4 个单电子,本文中的碳则是将基态的2 个处在不同2p 原子轨道中的2p2 电子,归并到一个轨道形成有2 对孤对电子,然后空出2 个空轨道,作为配位键的受体[9]。 由于电子成对要克服成对能,故此过程是能量升高的,导致这类配位键的稳定性下降[10]。因此,所研究的这一体系H3N→C←NH3、 (CH3)N→C←N(CH3)、H3P→C←PH3、(CH3)P→C←P(CH3),即碳的配合物体系(碳是受体),是一种新的发现,在催化过程中很重要。用7 种DFT 和G4 计算的8 种碳作为受体的配合物的配位键的键解离能在表1 给出。从表1 可以看出,与参考标准值G4 相比,7 种DFT 方法误差都不大,其中3 甲基磷与碳(G)形成的配位键的解离能最大,该类配合物也最稳定。但与传统的C-N 及C-P 键相比(对应的键解离能:C-P 513.8 kJ / mol,C-N 770.4 kJ / mol,CH3-NH2331.13 kJ / mol)均要小得多[11]。

表1 不同的DFT 与G4 方法计算的键解离能/(kJ·mol-1)
以这些特殊的配位键为例,对不同DFT 的适用性做一评价:
1) 对H3N→C←NH3而言,脱掉1 个-NH3的解离能误差最小的是BMK 方法。再解离另1 个-NH3,误差最小的DFT 方法是M06-2X。 把H 换成-CH3,即配合物CN(CH3)3-N(CH3)3,CN(CH3)3-N(CH3)3的解离能误差最小的方法是B2PLYPD。C-N(CH3)3的解离能误差最小的方法是M06-2X。从表1 可以看出,与参考标准值G4 相比,7 种DFT 方法误差都不大,均小于10 kJ / mol。
2) 用磷替换氮,对H3P→C←PH3而言,脱掉1个-PH3的解离能误差最小的是B3 方法。再解离掉1 个-PH3,最好的DFT 方法是M06-2X。 把H换成-CH3,即配合物CP(CH3)3-P(CH3)3,CP(CH3)3-P(CH3)3的误差最小的方法是B2PLYP。C-P(CH3)3的解离能误差最小的方法是B2PLYPD。
3) C 与1 个NH3结合是一能量升高的过程,导致解离时出现放热现象,于是无论何种DFT 方法计算均出现负的键解离能;这也说明即使形成N→C 配位键,其键能的释放也抵消不了处于不同原子轨道中自旋平行电子进入同一轨道克服排斥作用的成对能。只有再结合1 个NH3形成N→C 配位键,才能抵消成对能,导致总能量下降。
4) 7 种DFT 给出的MAD 在7.95 ~32.22 kJ/mol之间。M06-2X 给出的结果好,为7.95 kJ / mol。表现最差的泛函是B3LYP,其值为32.22 kJ / mol。
3 结论
通过上述 B3LYP,B2PLYPD,B2PLYP,B3,BMK,M05-2X,M06-2X 的密度泛函理论对新型C受体配合物的键解离能计算,发现应用这些方法结合选取较大基组可以得到较为满意的结果,于目前公认最高精度的G4 值较为一致。
从在7.95 ~ 32.22 kJ / mol 之间MAD 数据看,与解离能数据比较相对值大多数在1% 范围内,M06-2X 给出的结果好,为7.95 kJ / mol。即使表现最差的泛函B3LYP,其值为32.22 kJ / mol,相对值也小于10%。这些都表明上述方法对研究类似体系是可行的。
[1]徐文国,张利. CH3S,CH3SH,CH3SO,CH3SO2的标准摩尔生成焓的研究[J]. 北京理工大学学报,2004(09):825 - 828.
[2]苏欣纺,陈恒杰,程新路. 三硝基甲烷键离解能和生成焓的理论计算[J]. 原子与分子物理学报,2006(04):721 - 724.
[3]邵菊香,程新路,杨向东,等.C-H,C-N,C-O,N-N 的键解离能和键长的计算[J].原子与分子物理学报,2006(01):80-84.
[4]刘述斌. 概念密度泛函理论及近来的一些进展[J]. 物理化学学报,2009(03):590 - 600.
[5]宋晓书,令狐荣锋,葛素红. 含硝基烷基的一些CHNO 多硝基化合物的C-NO2键离解能计算-应用于撞击感度[J]. 原子与分子物理学报,2006(04):762 - 766.
[6]李震宇,贺伟,杨金龙. 密度泛函理论及其数值方法新进展[J]. 化学进展,2005(02):192 - 202.
[7]Robin A Y,Fromm K M.Coordination polymer networks with O-and N-donors[J].Coord.Chem Rev,2006,250(7):2127-2157.
[8] Mattsson A E. Density Functional Theory in Pursuit of the Divine Functiona [J]. Science,2002,25(10):759 - 760.
[9]Sheldrick G M. SHELXS-97 and SHELXL-97 Program for X-ray Crystal Structure Determination [D]. Germany:University of Gottingen,1997:120 - 125.
[10]Cole-Hamilton D J,Bruce D W,Wilkinson C.McCleverly Eds.In Comprehensive Coordination Chemistry [M].London:Pergamon Press,1987.
[11]Schilt A A. Analytical Applications of 1,10-Phenanthronline and Related Compounds [M]. London:Pergamon Press,1969.
