生长激素对急性坏死性胰腺炎大鼠肠粘膜损害NF-κB表达的影响*
2013-01-03武警陕西省总队医院胃肠外科西安710054李会文李向阳王周勤
武警陕西省总队医院胃肠外科(西安710054) 卢 翔 段 炜 张 力 李会文 李向阳 王周勤
急性坏死性胰腺炎(Acute necrotizing pancreatitis,ANP)肠道粘膜损伤的本质是由一系列炎症因子或介质介导的过度炎症反应和炎症损伤,可继发肠道屏障功能衰竭,肠腔内细菌及毒素移位,并进一步激发全身炎症反应综合征(Systemic inflammatory response syndrome,SIRS),肠道作为SIRS的原动力,ANP发生后NF-κB可活化并产生大量炎症介质,加剧肠道粘膜的损伤[1,2],这些促炎性细胞因子、趋化因子、粘附因子等基因的活化和表达表明核因子κB(NF-κB)的活化可能是其中的关键因素;有报道生长激素(Growth hormone,GH)具有维护ANP肠粘膜屏障功能,促进肠上皮修复,减少肠道细菌和毒素移位作用[3]。本实验通过观察ANP肠粘膜绒毛细胞(IMN)中 NF-κB的 表达 情 况,初步探讨GH对ANP肠粘膜NF-κB活化及其介导炎症介质表达的影响。
材料与方法
1 实验分组及急性坏死性胰腺炎(ANP)模型制备健康成年雄性SD大鼠36只,体质量250~300g,购自第四军医大学动物实验中心。以数字表法将动物随 机 分 成 三组:正常对照组、ANP组及GH处理组(处理组)。ANP模型制备:大鼠术前禁食12h,不禁水。20% 乌拉坦溶液(0.5ml/kg)腹腔注射麻醉,正中切口入腹,于肝十二指肠韧带近肝门侧用小动脉夹暂时夹闭胰胆管,由十二指肠前壁进针,进入胰胆管内,夹闭胰胆管出口,以0.1ml/min匀速逆行注入5% 牛磺胆酸钠(1ml/kg),10min后去除动脉夹,关腹。处理组在ANP模型制成前30min经尾静脉注射GH(0.75U/kg)。正常对照组自胰胆管内逆行注入等量生理盐水。
2 观察指标及实验方法
2.1 取材:末段回肠粘膜绒毛灌洗、细胞留取及各组动物手术后6h处死,然后取材。所有标本均重复检查2次。在动物处死后行末端回肠灌 洗,收 集 灌 洗液(TILF)。留取少量灌洗液,用于测定肿瘤坏死 因 子 -α(TNF-α)、白 细 胞 介 素1β(IL-1β)、诱导型一氧化氮合酶(iNOS)、细胞间粘附分子(ICAM-1)和单核细胞趋化蛋白(MCP-1)。将 剩余灌洗液离心获取细胞沉淀,备用。大鼠末端回肠用于组织学检查。
2.2 病理形态学检查:将各组大鼠的末端回肠置10% 甲醛溶液中浸泡,石蜡包埋,行苏木精 -伊红(HE)染色,光学显微镜下观察回肠组织学改变。
2.3 BALF中肿 瘤 坏 死 因 子 -α(TNF-α)、白细 胞 介 素1β(IL-1β)、诱导型一氧化氮合酶(iNOS)、细胞间粘附分子(ICAM-1)和单核细胞趋化蛋白(MCP-1)测定:应用 BioSoure公 司 的 TNF- α、IL-1β、iNOS、ICAM-1和MCP-1酶 联 免 疫 吸 附法(ELISA)试剂盒测定,严格按照说明书操作。
2.4 Western blot检 测 NF-κB含 量:应用北京普利莱公司胞核 -胞浆蛋白制备试剂盒提取肠粘膜组织核蛋白并测定浓度后,行聚丙烯酰胺凝胶电泳。结果行计算机扫描,用图像分析系统,测 定 目 的 条 带 与内 参 的 光 密 度 值(OD)。并取两者的比值作半定量分析。
3 统计学方法 数据以均数 ± 标准差表示。数据间比较采用单因素方差分析,相关性采用线性分析,应用SPSS13.0分析软件分别对各组的均数行统 计学处理。P<0.05认为差异有统计学意义。
结 果
1 肠粘膜组织大体变化及病理组织学所见:对照组大鼠肠粘膜呈粉红色,表面光滑,无渗出及出血;光学显微镜下见肠粘膜绒毛组织结构清晰,肠粘膜细胞壁薄,肠粘膜内未见渗出液。ANP组肉眼见整个肠粘膜充血,间有斑片状坏死,镜下见肠粘膜间质水肿,弥漫性出血,肠粘膜细胞结构紊乱,血管扩张淤血,有大 量 炎性细胞浸润,肠粘膜内有炎性渗出。GH处理组上述变化较ANP组明显减轻(图1)。

图1 各组肠粘膜组织病理切片(HE×200) A:ANP组;B:对照组;C:GH处理组(图片A、C中,箭头所指为细胞病理改变)
2 肠粘膜组织中 TNF-α、IL-β、iNOS、ICAM-1和MCP-1含量的变化:正 常 大 鼠肠粘膜组织中TNF-α、IL-β、iNOS、ICAM-1和 MCP-1含量 较 低,处 理 组 与正常对照组相比,各指标差异均无统计学意义;ANP组各项指标均高于正常对照组和处理组,差异有统计学意义(P <0.05),见表1。
表1 肠粘膜组织中 TNF-α、IL-β、iNOS、ICAM-1和 MCP-1含量 (±s)
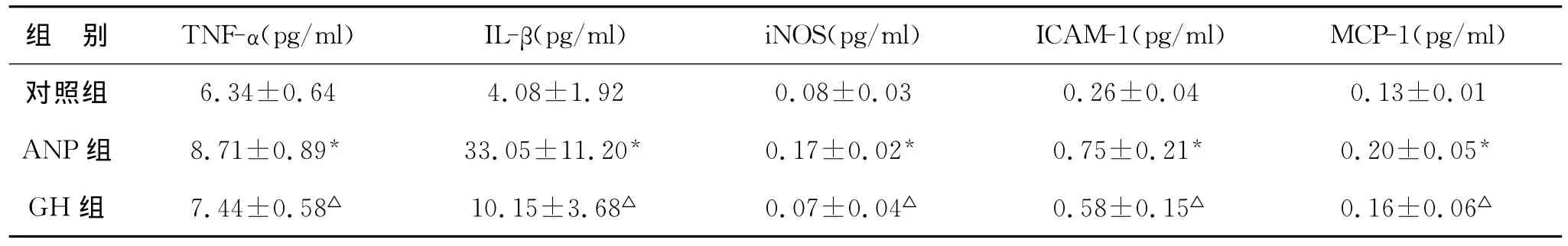
表1 肠粘膜组织中 TNF-α、IL-β、iNOS、ICAM-1和 MCP-1含量 (±s)
注:*与对照组比较P<0.05;△与ANP组比较P<0.05
组 别 TNF-α(pg/ml) IL-β(pg/ml) iNOS(pg/ml) ICAM-1(pg/ml) MCP-1(pg/ml)对照组 6.34±0.64 4.08±1.92 0.08±0.03 0.26±0.04 0.13±0.01 ANP组 8.71±0.89* 33.05±11.20* 0.17±0.02* 0.75±0.21* 0.20±0.05*GH组 7.44±0.58△ 10.15±3.68△ 0.07±0.04△ 0.58±0.15△ 0.16±0.06△
3 各组肠粘膜绒毛核蛋白中NF-κB P65活性:NF-κB P65在 对 照 组 的肠粘膜绒毛核 蛋 白 中 表 达程度最低,在ANP组中呈高表达,在GH处理组呈中度表达(图2),见表2。
4 相关性分析:在ANP组,肠粘膜组织中NF-κB P65的表达呈高度相关(r=-0.706,P =0.015);在 GH处理组,肠粘膜组织中NF-κB P65的表达呈中度相关(r=-0.572,P =0.019)。
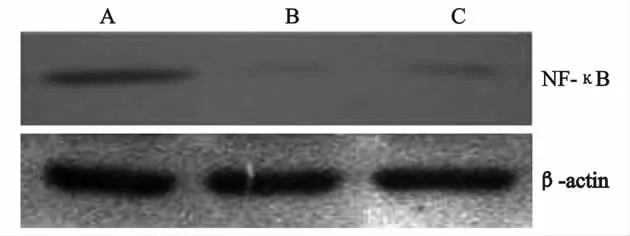
图2 各组肠粘膜绒毛核蛋白中的NF-κB表达
表2 肠粘膜组织核蛋白中NF-κB P65/β-actin相对量(±s)

表2 肠粘膜组织核蛋白中NF-κB P65/β-actin相对量(±s)
注:*与对照组比较P<0.05;△与ANP组比较P<0.05
组 别 NF-κB P65/β-actin对照组0.08±0.03 ANP组 0.18±0.06*GH组 0.11±0.04△
讨 论
急性坏死性胰腺炎可激发全身炎症反应综合征的发生,肠道受缺血、缺氧等因素的刺激,在肠粘膜绒毛细胞合成大量细胞因子等炎症介质,一方面加重肠道屏障功能的损害、衰竭,另一方面则促使全身炎症反应不断扩大[4]。目前有研究报道,核因子-κB(Nuclear factorκB,NF-κB)是调控多种炎性因子基因表达的枢纽,是一重要的细胞内信号传导物质,活化后启动细胞核内相应的细胞因子、粘附分子和趋化因子的启动子,调节炎症和免疫反应[5]。本实验结果发现,ANP组IMN中NF-κB表达活性明显高于正常对照组及处理组;TNF-α、IL-β、iNOS、ICAM-1和 MCP-1的 表 达 较 正 常 对 照 组及处理组明显升高。提示在ANP时有NF-κB的大量激活,导致多种炎症介质的过量释放,使炎症反应持续加重,进一步加重肠粘膜损伤。近来发现生长激素(GH)具有维护ANP肠粘膜屏障功能,促进肠上皮修复,减少肠道细菌和毒素易位的作用。提示GH可防止肠上皮细胞过度凋亡,下调炎症介质的释放,具有抗炎效应[6]。GH具有促进蛋白质合成,促进烧伤、创伤、感染等患者正氮平衡的恢复和创面组织生长,同时还可维持肠粘膜正常结构,促进肠粘膜功能的恢复[7];本研究观察发现GH下调它们的表达,提示GH可能通过降低TNF-α、IL-β、iNOS、ICAM-1和 MCP-1的表达影响局部白细胞浸润,从而减轻肠粘膜屏障的炎性损伤。
核转录因子NF-κB是由可诱导的3个亚基组成的复合物,3个亚基分别为转录因子二聚体(P50和P65)和抑制亚基Ⅰ-κB。NF-κB由于ⅠκB的抑制作用主要以非活性形态存在于细胞质。转录因子NF-κB在许多领域受到研究者的关注基于以下的几点:罕见和快速的调节性,控制的基因范围广,在免疫过程处于中枢的角色,亚基的复杂性和牵涉若干个疾病。控制NF-κB的基本水平是通过与抑制蛋白ⅠκB的相互作用。最近的证据证实了不同机制调节 NF-κB的IκB多样形态存在。NF-κB可以被脂肪酶、脂多糖或者炎症细胞因子激活例如TN F、IL-1、病毒感染、某一个病毒基因产物的表达、紫外线照射、B或T细胞活化、其他生理学的和非生理学的刺激物。NF-κB激活转移至细胞核是通过ⅠκB定向磷酸化、降解,与NF-κB解离进行的。NF-κB一旦转入细胞核即结合同族DN A序列和激活特异靶基因转录,大多数是编码免疫和炎症的重要蛋白。NF-κB/Rel因子控制大范围的基因表达,例如编码免疫球蛋白、细胞因子、化学增活素、干扰素、主要组织相容性复合物蛋白质、生长因子、细胞粘附分子。NF-κB可与许多基因的启动子结合,包括炎症应答例如ICAM-1、E-选择蛋白、VCAM、TNF-α、IL-1、IL-2、IL-6、IL-8、COX2 和iNOS[8~13]。因此,核转录因子NF-κB是炎症反应的关键角色。本研究用P65单抗结合伊红复染细胞浆检测大鼠肠粘膜细胞核内P65亚单位,正常状态P50/P65复合体只存在于细胞浆,因此该方法可以观察NF-κB的活化情况。结果观察发现对照组肠粘膜阳性细胞数很少,而ANP组可发现大量NF-κB P65阳性细胞数;说明在ANP早期肠粘膜NF-κB就大量活化,介导了炎症反应的扩大。NF-κB P65阳性细胞主要为肠绒毛顶端的上皮细胞及其下面的单核细胞,可能因为肠绒毛顶端作为接触毒素的最前沿,因而该处的炎症反应也最强烈。
本研究发现GH处理组NF-κB P65阳性细胞较ANP组明显减少,说明GH有抑制NF-κB活化作用。结合GH能显著下调ANP肠组织NF-κB介导的细胞因子表达,从而进一步揭示NF-κB及其介导的细胞因子表达参与了ANP肠道粘膜的损伤的发生。GH对ANP肠道的保护作用与GH下调肠粘膜细胞因子表达、抑制NF-κB活化有关。
[1] Shimada M,Andoh A,Hata K,et al.IL-6secretion by human pancreatic periacinar myofibroblasts in response to inflammatory mediators[J].J Immunol,2002,168(2):861-868.
[2] Seo SW,Jung WS,Lee SE,et al.Effects of bee venom on cholecystokinin octapeptide-induced acute pancreatitis in rats[J].Pancreas,2008,36(2):e22-29.
[3] Wang X,Wang B,Wu J,et al.Beneficial effects of growwth hormone on bacterial translocation form gut during the course of acute necrotizing pancreatitis inrats[J].Pancreas,2001,23:148-156.
[4] Ogawa M.Acute pancrentitis and cytokines:“second attack”by septic complication leads to organ failure[J].Pancreas,1998,16:312-315.
[5] Neuracb MF,Becker C,Barbulescu K.Role of NF-kB in immune and inflammatoryresponse in the gut[J].Cut,1998,43:856-860.
[6] 王兴鹏,王冰娴,吴 恺,等.急性坏死性胰腺炎肠粘膜NF-kB介导的细胞因子过度表达及生长激素的作用[J].中华肝胆外科杂志,2003,9(1):45-49.
[7] 顾 军,黎介寿,李 宁,等.生长激素对腹腔感染后肠粘膜结构和细胞增生影响的研究[J].医学研究生学报,2003,19(1):13-16.
[8] Chen X,Ji B,Han B,et al.NF-kappaB activation in pancreas induces pancreatic and systemic inflammatory response[J].Gastroenterology,2002,122:448-457.
[9] Rakonczay Z Jr,Jarmay K,Kaszaki J,et al.NF-kappaB activation is detr.imental in arginine-induced acute pancreatitis[J].Free Radic Biol Med,2003,34:696-709.
[10] Baldwin AS Jr.The NF-kappa B and I kappa B proteins:new discoveries and insights[J].Annu Rev Immunol,1996,14:649-683.
[11] Chen Z,Hagler J,Palombella VJ,et al.Signal-induced site-specific phosphorylation targets I kappa B alpha to the ubiquitin-proteasome pathway[J].Genes Dev,1995,9:1586-1597.
[12] Verma IM ,Stevenson JK,Schwarz EM .Rel/NF-kappa B/I kappa B family:intimate tales of association and dissociation[J].Genes Dev,1995,9:2723-2735.
[13] Li X,Massa PE,Hanidu A,et al.IKKalpha,IKKbeta,and NEMO/IKKgamma are each required for the NF-kappa B-mediated inflammatory response program[J].J Biol Chem,2002,277:45129-45140.
