硫解高效液相色谱法快速检测原花青素
2012-12-23潘玉雷
侯 栋,李 晔,王 洋,潘玉雷,崔 同
河北农业大学食品科技学院,保定071000
原花青素(proanthocyanidins,简称PC)是一种能在酸性条件下加热分解产生花色素的缩合多酚类次生代谢产物,存在于山楂、葡萄、松树、高粱、苹果、红景天、可可、白桦等多种药用、食用植物组织中。大量研究表明,PC 是一种性能优异的天然抗氧化剂和自由基清除剂[1,2],具有保护心脑血管、抗炎、抗辐射、降低癌症风险等一系列重要生理作用,是一种重要的天然产物资源。
PC 常以黄烷-3-醇类为主要结构单元,通过4→8 位或4→6 位连接,聚合成十分复杂的混合物,不同植物来源、键合方式、聚合程度、加工提取方法等因素都会影响PC 产品的组成和含量,从而影响产品的保健功效。因此,如何准确客观地表征和分析PC 的组成和含量,一直是制约PC 产业标准化的重要问题。
目前PC 行业使用最广泛的分析方法是分光光度法,如香草醛-盐酸法[3]、Bate-Smith 法[4]、Porter法[5]等。这些方法包括目前国标方法的缺陷在于,以某种指定对照品进行的分析,其结果不能准确反映不同来源PC 含量的真实差异。HPLC 法[6]包括液谱-质谱联用法(LC-MS)[7]虽然能对PC 的主要成分直接进行分离和定量分析,但由于PC 组成十分复杂,目前除几种小分子PC 成分之外,其余PC 纯品很难从市场上获得,并且HPLC 法也很难对所有PC 成分实现彻底分离。
Thompson 等[8]曾发现,在有亲核试剂例如苄硫醇存在下,PC 可在酸性条件下热分解,基部单元生成相应的黄烷醇分子,上部的其余单元生成相应的苄硫醚衍生物。这便可以使组成十分复杂的PC 混合物经硫解反应后只生成有限几种反应产物。通过分析这些产物的含量不仅可以获得样品中PC 总含量的结果,而且可以通过苄硫醚衍生物总含量与黄烷醇含量的比例关系,计算出样品中PC 成分的平均聚合度。目前这种方法多应用于PC 成分的结构鉴定,以及一些初步的HPLC 定量分析法[9]。本项研究拟在此基础上,通过制备和纯化PC 的苄硫醚衍生物,并以此为对照品,通过优化色谱条件,开发一种新型的硫解-HPLC 快速分析方法,以便实现PC含量和平均聚合度的同时检测,为PC 类植物提取物的产品质量评价提供更可靠的分析手段。
1 材料与方法
1.1 仪器
定量分析用HPLC:Agilent 1200 型(安捷伦公司,美国加州帕罗阿托市),由在线脱气机,低压梯度四元输液泵,Agilent Chemstation 色谱工作站,光电二极管阵列检测器,CO-3010 柱恒温控制箱(天津美瑞泰克科技有限公司生产),及Hypersil BDS C18(100 mm × 2.1 mm i.d.,3 μm)色谱柱组成。
制备HPLC(大连依利特科学仪器有限公司,大连):由P260 型输液泵,紫外-可见检测器,Echrom 2000 色谱工作站组成,配用SinoChrom ODS-BP C18(20.0 mm × 300 mm id,10 μm )以及SinoChrom SiO2(20.0 ×250 mm id,10 μm)制备色谱柱。
LC-MS 系统:LTQ 型,美国赛默飞世尔(Thermo Fisher)科技有限公司生产。
1.2 材料
葡萄籽提取物(标示含量>95%),陕西中鑫生物技术有限公司生产。山楂,2011 年6 月23 日采自河北农业大学标本园。
1.3 试剂
没食子酸(gallic acid,简称GA),表儿茶素(epicatechin,简称EC),儿茶素(catechin,简称CT)购于Sigma 公司。苄硫醇购于Fluka(Buchs,Switzerland)公司;表儿茶素没食子酸酯(epicatechin gallate,简称ECG)购于上海同田生物科技有限公司。色谱级甲醇和乙腈购于Honeywell Burdick & Jackson 公司。表儿茶素-4-苯甲硫醚(EC-S)和表儿茶素没食子酸酯-4-苯甲硫醚(ECG-S)按下列方法制备:取葡萄籽提取物2 g 溶解于3 mL 甲醇,吸取500 μL 上清液,加入500 μL 含30%苄硫醇的乙醇溶液和250 μL 冰乙酸:1M 盐酸(50:1,v/v)溶液混合,密封后在90℃反应60 min。然后将硫解产物混合液用反相制备HPLC 反复进样分离纯化,色谱柱为SinoChrom ODS-BP C18(300 mm×20 mm id,10 μm ),流动相为70%甲醇(含0.05%甲酸),流速10 mL/min,在280 nm 波长下检测流出物,收集目标馏分于40 ℃减压浓缩除去甲醇,再用乙酸乙酯萃取,减压浓缩。然后用SinoChrom SiO2(250 ×20 mm id,10 μm)正相柱纯化,流动相:乙酸乙酯∶石油醚(60∶40,v/v),流速10 mL/min,检测波长280 nm,收集目标馏分浓缩至干,最后得到两种硫醚衍生物纯品。经LC-MC 分析,其质谱信息与EC-S 和ECG-S 的结构吻合(见图2),其色谱保留行为与文献报道一致。其余试剂为分析纯。
1.4 样品前处理
取山楂青果,去籽去梗后切碎混匀,精确称取5 g 至研钵,加入20 mL 95%乙醇溶液(含0.2% Vc,0.2%磷酸)后研磨至粥状,转移至50 mL 容量瓶中,超声提取10 min,用70% 乙醇(含0. 2% Vc,0.2%磷酸)定容至50 mL,混匀、静置后离心(4000 rpm,10 min),取上清液过0.45 μm 微孔滤膜,备用。
葡萄籽提取物混匀后精确称取10 mg,用95%乙醇溶解定容至10 mL,取1 mL 液体用0.45 μm 微孔滤膜过滤,备用。
1.5 硫解反应
反应A 液:5%(v/v)苄硫醇乙醇溶液;反应B液:冰醋酸∶1M 盐酸(50∶1 v/v)。取100 μL A 液,50 μL B 液,100 μL 样品溶液,放入小瓶中密封,90℃下水浴反应60 min 后立即冷却,用于HPLC 分析。
1.6 HPLC 色谱条件
流动相A:0.05%甲酸水溶液;流动相B:乙腈∶甲醇(2∶1,v/v);梯度洗脱程序(min/B%):0/6%,4/50%;6/55%;7/100%;8/100%;9/6%;15/6%;柱温:35 ℃;流速:0.2 mL/min;进样量:1 μL;检测波长:280 nm;定量:峰面积外标法。
1.7 质谱条件
正离子扫描(ESI,m/z 50~1000);毛细管电压:33 V;光电倍增器电压:-1560 V;补偿电压:95 V;离子源温度:35 ℃;不分流检测。
2 结果与讨论
2.1 硫醚衍生物对照品的LC-MS 分析
采用1.6 和1.7 所述的LC-MS 方法对这两种硫醚衍生物进行了阳离子模式分析,图1 示这两种成分的阳离子质谱图。

图1 两种硫解产物正离子模式质谱图Fig.1 Mass spectrums of two Thiolysis Derivatives
图1 中(A)给出了质荷比413 的质谱峰,与ECS 的分子离子[M + H]+一致,质核比289 的质谱峰与EC 碎片离子[M–C7H8S]+一致。因此,质谱分析结果支持其结构为EC-S 的判断。同理,(B)图给出了质核比为565 的质谱峰,它与ECG-S 的分子离子[M + H]+一致,而质核比为441 的质谱峰与ECG-S 分子去掉硫醚后的ECG 碎片离子[M –C7H8S]+的质量一致,质谱分析结果支持其结构为ECG-S 的判断。
2.2 对照品贮备液的配制
分别准确称取对照品GA、CT、EC、ECG、EC-S和ECG-S,用甲醇配制成2 mg/mL 的贮备液,使用前取贮备液用50%甲醇配制相应浓度的混合对照品溶液。
2.3 方法精密度
取2.2 所述对照品贮备液配制混标液,使其中GA、CT、EC、ECG、EC-S 和ECG-S 的浓度分别为160、480、320、160、480 和320 μg/mL,用50%甲醇稀释1 倍后按1.6 所述HPLC 方法重复进样5 次,测定峰面积,以评价方法的精密度。计算得6 种对照品峰面积RSD 分别为1.37%、1.09%、1.60%、0.74%、1.85%和1.86%。结果表明,该方法精密度良好。
2.4 线性关系与检出限
将混合对照品溶液用50% 甲醇逐级稀释,按1.6 所述HPLC 方法进样分析,测定峰面积,以峰面积(mAU·S)为纵坐标,以其进样量(ng)为横坐标,绘制标准曲线,并以3 倍信噪比(S/N)时的进样量确定最低检出限。求得6 种对照品的线性回归方程、线性回归系数、最低检出限,结果见表1。结果表明,响应值与进样量之间具有良好线性相关性,线性范围至少320 倍,最低检出限在ng 数量级,可以满足常规样品的定量分析。
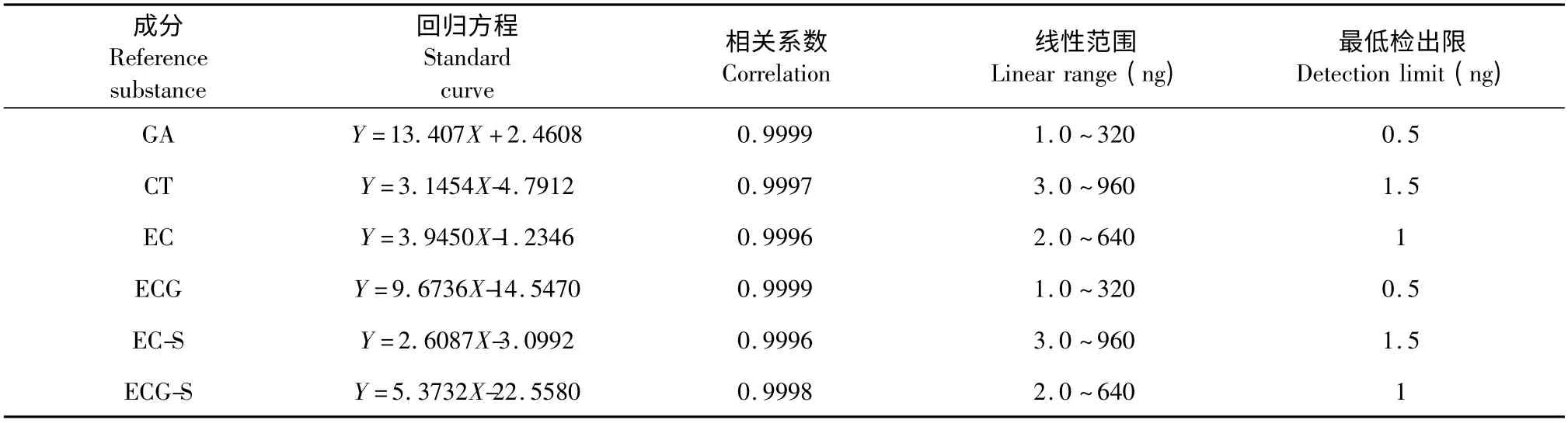
表1 对照品的回归方程、相关系数、线性范围及最低检出限Table 1 Regression equation,correlation coefficient,linear range and the minimum detection limit
2.5 加标回收率分析
采用加标回收率法对方法的准确度进行了评估。重复精确称取葡萄籽提取物样品40 mg,溶于10 mL 甲醇溶液中,按照1.5 所述的硫解衍生反应进行处理,再按1.6 所述的方法逐个进样分析,统计各成分的分析结果,计算各成分含量的平均值。然后另取5 份样品,每份中均添加含有GA、CT、EC、ECG 的混合对照品,四种成分的加标水平分别为5,65,20 和10 mg/g(干重)。按照1.5 所述的硫解衍生反应进行处理,再按1.6 所述的方法逐个进样分析,统计各成分的分析结果,计算各成分的加标回收率及其RSD 值,结果见表2。由表中可以看出,CT、EC、ECG 的回收率较高,而GA 的平均回收率偏低,推测GA 比较活泼,在衍生反应过程中存在某种影响因素。
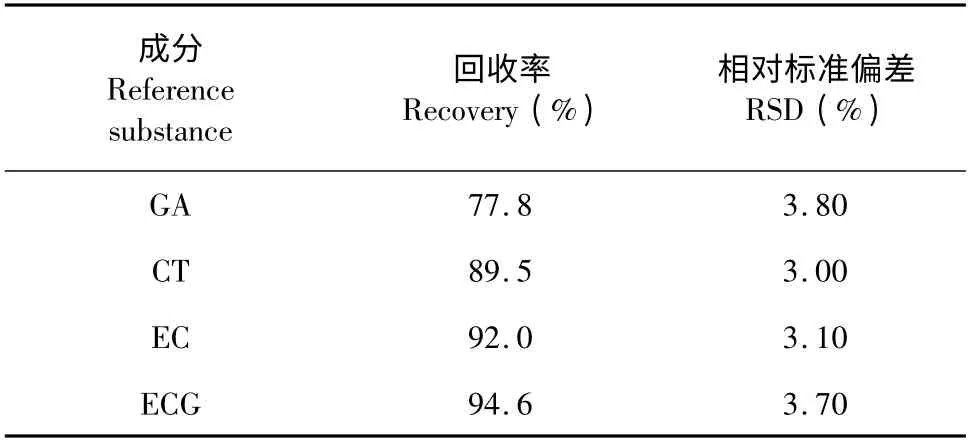
表2 各成分回收率Table 2 Recoveries of gallic acid,catechin,epicatechin and epicatechin gallate
2.6 样品分析
将混合对照品液按1.6 所述方法进样HPLC 分析,实际样品按1.4 所述方法进行样品前处理,按1.5 所述方法进行硫解并按1. 6 所述方法进行HPLC 分析,典型的色谱图如图2 所示。从图2(A)可以看出,6 种对照品可在11 min 以内完成分离,比以往的方法更快捷。从图2(B)图2(C)中可以看出,葡萄籽提取物和山楂样品经过硫解反应之后色谱图变得非常简单,除本试验选用的6 种主要对照品之外,其它只在8 min 和10 min 之间有3 个含量不高的未知成分,而原来聚集在6~10 min 处的高分子聚合物杂质的大峰已基本消除,这使得PC 的定量分析结果变得更加准确可靠,不同植物来源的PC 之间的比较也变得更加简单可信。结果表明,不同植物来源的PC,其单体组成差异很大,例如山楂果实的PC 中不含GA、CT 和ECG,因此图谱显得更加简单。

图2 对照品(A)、山楂硫解样品(B)及葡萄籽提取物硫解样品(C)的HPLC 图Fig.2 HPLC profiles (detected at 280 nm)of reference standards (A),HPLC traces of polymeric proanthocyanidins from immature Chinese hawthorn (B)after thiolysis and HPLC traces of polymeric proanthocyanidins from grape seed extracts after thiolysis (C)including gallic acid,flavan-3-oL and their relational thiolysis derivatives
两份实际样品经过硫解-HPLC 分析,代入相应的标准曲线求得结果如表3。从表3 可以看出,山楂青果中其PC 含量很高,为38.42 mg/g。葡萄籽提取物中PC 含量为427.2 mg/g。
2.7 平均聚合度的计算
根据PC 硫解反应原理[8]可知,样品经过硫解反应后,其末端生成黄烷醇单体,而上部的其余各单元均生成相应的苄硫醚衍生物。因此,以GA、CA、EC、ECG、EC-S、ECG-S 这些化合物为对照品,可以对样品中所含原花色素总量进行测定。样品中PC的平均聚合程度(mDP)则可以由硫解反应液中生成的所有黄烷醇单体的总浓度除以所生成的末端黄烷醇单元浓度之比来计算。即:按以下公式计算:

表3 实际样品PC 含量(mg/g)Table 3 Contents of PC constitutes in two tested samples
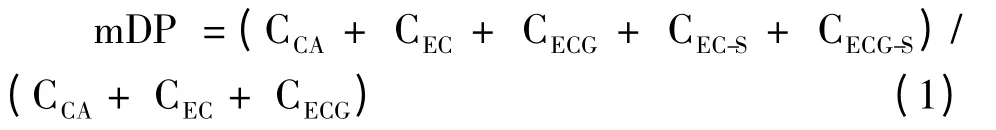
(1)式中:
CCA——儿茶素的摩尔浓度;
CEC——表儿茶素的摩尔浓度;
CECG——表儿茶素没食子酸酯的摩尔浓度;
CEC-S——表儿茶素苄硫醚衍生物的摩尔浓度;
CECG-S——表儿茶素没食子酸酯苄硫醚衍生物的摩尔浓度。
由表3 的分析结果计算得出各成分的摩尔浓度,带入式(1)即可求出样品中PC 的平均聚合度。山楂青果和葡萄籽提取物的平均聚合度分别为2.97 和4.83。有研究结果表明,只有PC 单体和二聚体能够被人体小肠的细胞吸收,而三聚体以上的聚合PC 一般不能被人体直接吸收,因此,PC 的平均聚合度是评价PC 质量的一个重要指标。
3 结论
研究结果表明,采用本研究的苄硫醇酸解条件可以使样品中的聚合PC 分子降解为黄烷醇单体或相应的硫醚衍生物,再用HPLC 法实现了6 种硫解产物在11 min 内的快速分离,15 min 完成一次样品分析,方法具有良好的灵敏度、精密度和准确度。这种方法的另一个优点是,在提供PC 总含量结果的同时还能给出样品中PC 成分平均聚合度的重要信息,为PC 产品质量评价提供了新的分析手段。本项研究的另一个特点是制备纯化了两种黄烷醇的硫醚衍生物并以此作为HPLC 分析的外标对照品。本方法与以往的分光光度法原理不同,两者的分析结果可能会有差异,这方面还需要开展大量的分析验证研究。
1 Lv LS(吕丽爽),Cao D(曹栋).Study on the antioxidant activity of oligermeric proanthocyanidins.Food Sci Technol(食品科技),2000,4:41-42.
2 Zhu JR(朱婧蓉),Wang ZM(王忠民),Li JY(李谨瑜),et al.Study on the antioxidant activity of oligermeric proanthocyanidins.Food Nut Chin(中国食物与营养),2007,4:34-37.
3 Yao K(姚开),He Q(何强),Lv YP(吕远平),et al.Determination of proanthocyanidin from grape-seed extracts. J Food Fer Ind(食品与发酵工业),2002,28(3):17-19.
4 Bate-Smith EC. Phytochemistry of proanthocyanidins. Phytochemistry,1975,14:1107-1113.
5 Porter LJ,Hrstich LN,Chan BG.The conversion of procyanidins and prodelphinidins to cyanidin and delphindin.Phytochemistry,1986,25:223-230.
6 Svedstrom U,Vuorela H,Kostiainen R. High-performance liquid chromatographic determination of oligomeric procyanidins from dimers up to the hexamer in hawthorn.J Chromatogr A,2002,968:53-60.
7 Fulcrand H,Remy S,Souguet JM. Study of wine tannin oligomers by on-line liquid chromatography electro-spray ionization mass spectrometry. J Agric Food Chem,1999,47:1023-1028.
8 Thompson RS,Jacques D,Haslam E,et al.Plant proanthocyanidins.part I. introduction;the isolation,structure,and distribution in nature of plant procyanidins. JCS Perkin I ,1972,1387-1399.
9 Morimoto S,Nonaka G-I,Nishioka I. Tannins and related compounds. XLI. Isolation and characterization of novel ellagitannins,punicacorteins A,B,C,and D,and punigluconin from the bark of Punica granatum L. Chem Pharm Bull,1986,34:656-663.
