Li修饰B12N12储氢行为的密度泛函理论研究
2012-11-30许文杰胡自玉邵晓红
许文杰 胡自玉 邵晓红
(北京化工大学理学院,北京100029)
Li修饰B12N12储氢行为的密度泛函理论研究
许文杰 胡自玉 邵晓红*
(北京化工大学理学院,北京100029)
采用基于密度泛函理论(DFT)的第一性原理投影缀加波方法,研究了Li修饰的B12N12笼子的储氢行为.计算结果表明:Li原子吸附在B12N12笼子的四元环和六元环相交的B-N桥位上,相对于其它六个高对称吸附位置更稳定,B12N12笼子周围最多可以吸附3个Li原子,最稳定的构型是三个Li原子同时吸附在N原子顶位(Top-N site).每个Li原子的周围能吸附三个氢分子,笼子外侧还可以吸附两个氢分子,内部最多可以吸附5个氢分子.考虑到笼内和笼外的吸附,B12N12笼子总的储氢量(氢分子)达到9.1%(w).
第一性原理;修饰;B12N12;储氢;吸附能
1 引言
Golberg1,2、Ma3、Oku4-7等用强电子束在高温下照射氮化硼发现孤立的单层空心正八面体的BN笼状结构以来,(BN)n因结构稳定、耐高温、宽带隙等特点受到广泛关注.Folwer等8使用Spiral算法生成了所有由四元环与六元环形成的(BN)n(n=4-30)结构,发现(BN)12具有Th的对称结构,是包含四元环和六元环数目最小的多面体.Chattaraj等9用密度泛函理论(DFT)方法研究了nH2@B12N12(n=1-12)体系的稳定性,其中B12N12最多在表面可以吸附12个氢分子,折合储氢量为7.45%(w).该研究结果表明B12N12是有希望的储氢材料.
Li等金属原子修饰被视为有效提高储氢量的方法之一.10-17Sun等18通过DFT和分子动力学模拟(MD)方法研究了Li修饰的B36N36的储氢行为,研究结果表明Li修饰在B36N36上可以有效提高储氢量.此外,Wen10和Sun18等的研究结果表明氢气倾向于以分子形式吸附在纯的B36N36笼子内部,而关于Li原子修饰后氢气分子在B12N12笼内和笼外储存的系统研究未见报道.
本工作采用基于密度泛函理论的第一性原理软件VASP研究了Li掺杂的B12N12笼子的储氢行为.首先讨论了B12N12最多可以修饰的Li原子个数,以及单个和多个Li原子的最稳定吸附位.其次,讨论了氢气在笼子内部和外部的吸附行为,包括储氢量等,为类富勒烯结构材料的储氢提供了理论参考.
2 计算方法
本文计算工作采用基于密度泛函理论的第一性原理软件VASP,计算中使用投影缀加波方法(PAW)产生的赝势和平面波基组,交换关联势采用广义梯度近似的(GGA)PW-91势.B12N12笼子的弛豫是在x、y、z方向上各2 nm的真空层组成的超晶胞里完成的,从而忽略了笼子间的相互作用,保证了计算收敛的高效性.采用了3×3×3k点网络对布里渊区进行积分,平面波截断能设为400 eV.计算弛豫总能的收敛标准为1×10-5eV·nm-1,进行离子弛豫时用分子动力学模拟,收敛标准为各个原子之间的作用力小于1×10-4eV·nm-1.此外,测试结果发现体系的自旋磁矩为零,没有自旋极化的作用.
3 结果与讨论
3.1 B12N12结构和局域电荷分布
图1(a)是优化好的B12N12笼子结构图,其包含的12对B-N键均匀分布在6个四元环和8个六元环上.四元环和六元环共有的B-N键长为0.149 nm,而在六元环上的B-N键长为0.144 nm,此结果与之前的结论吻合得非常好.19图1(b)中给出了B12N12笼子的电荷局域分布,哑铃形状代表电荷为负,球状部分代表电荷为正.从图中可以很明显地看到,B原子周围是电荷缺失的地方,N原子周围是电荷增加的地方,即B-N键具有很强的离子性,正是由于B原子周围的电荷缺失也使得该位置成为氢原子最有利的吸附位置.图1(c,d)中给出了纯的B12N12的电子最高占据分子轨道(HOMO)和最低未占据分子轨道(LUMO)分布,可以清楚地看出B12N12笼子的最高占据分子轨道主要局域在N原子周围,而最低未占据分子轨道则主要局域在B原子周围,这也是B12N12笼子具有一定带宽的原因所在.从电荷局域分布上可以看出,在B和N原子上的杂化轨道类型分别近似为sp2和sp3类型.
3.2 Li在B12N12上的吸附

图1 B12N12的结构(a)、局域电荷密度(b)和前线分子轨道(c,d)Fig.1 Structure(a),local electronic density(b),and frontier molecular orbitals(c,d)of B12N12(c)the highest occupied molecular orbital(HOMO);(d)the lowest unoccupied molecular orbital(LUMO); ELF:electron localization function
首先讨论了单个Li原子与纯的B12N12相互作用.Li原子吸附在B12N12上7个高对称吸附位如图2所示.其中a和b表示四元环和六元环的中心位置,c和d代表B原子和N原子的顶端位置,e和f分别表示四元环和六元环相交的B-N桥位以及六元环与六元环相连的B-N桥位,g表示B12N12笼子的正中心位置.结构优化时,首先将Li原子放置在上述7个高对称吸附位上,弛豫后发现在a、b和c位置上的Li原子会分别跑到d、e和f位置上.Li原子的吸附能用如下公式进行计算:
Ead=ELiB12N12-ELi-EB12N12(1)其中,ELi表示单个Li原子的能量,ELiB12N12和EB12N12分别代表了LiB12N12笼子和B12N12笼子的总能量.吸附能越小,表明Li原子吸附的系统越稳定.按照公式
(1)计算得出的Li原子位于d、e、f和g的吸附能分别为-0.75、-0.76、-0.75和-0.34 eV,该结果表明位于四元环和六元环相交的B-N键上的桥位e是最稳定的吸附位置.
下面我们讨论多个Li原子的吸附.当多个Li原子吸附时可能出现成簇现象.11,18,20首先将两个Li原子放置在高对称吸附位d、e、f上,优化结果表明:当两个Li原子分别位于相邻的六元环上时,成簇的现象比较明显;当两个Li原子分别位于和一个四元环相交的另外两个相邻的六元环上的d、e、f稳定吸附位置时,Li原子之间距离为0.386 nm,即没有发生成簇现象.按照以上方式排布,B12N12周围只能容纳三个Li原子.为了验证这一结论,我们计算了4个Li原子吸附在B12N12周围的情形,确实发现有两个Li原子逐渐靠近,出现了成簇现象.因此,以下讨论中我们只考虑3个Li原子的情形,当3个Li原子同时放在d、e、f稳定吸附位置时,按照公式(2)

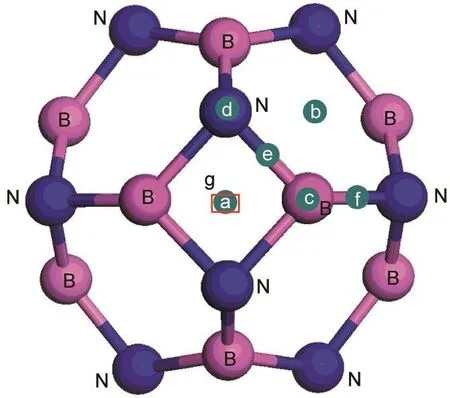
图2 B12N12笼子的七个高对称位Fig.2 Seven high symmetry sites of B12N12cage
计算得出平均吸附能分别为-2.89、-2.86、-2.84 eV.由此可以看出Li3B12N12的平均吸附能远小于LiB12N12,这与之前报道的Li6B36N36的体系的结果是一致的.10更有趣的是当Li原子全部吸附在d位置时具有的平均吸附能为-2.89 eV,远远高于主体Li原子的粘合能-1.70 eV,进而可以有效地阻止Li原子之间成簇.
图3(A)表示Li原子吸附在e位置上最稳定的构型图,Li原子与N和B所成的键长分别是0.189和0.213 nm,稍微偏向于N原子.图3(B)中给出了Li吸附在d位置上最稳定的结构,Li原子与N键长是0.189 nm.对比可以发现,单个Li原子吸附在桥位最稳定,但是当吸附到三个Li原子的时候,发现同时吸附在N原子顶位(Top-N site)时是最稳定的.因此,本工作以Li原子吸附在Top-N位置来讨论氢分子的吸附.图3(C,D)中给出了Li原子吸附在d位置之后的前线轨道图.结合图1(c,d)可以看出Li原子吸附之后,最高占据轨道主要分布在Li原子周围,如图3(C)所示,最低未占据轨道虽在Li原子周围有部分分布,但是底部的轨道排布改变并不明显.从图3 (D)可以看出Li原子吸附之后B12N12上面的电荷局域分布的杂化类型仍是sp3类型.
3.3 氢在Li修饰的B12N12上的吸附

图3 Li吸附在e和d位置上的最稳定的构型图以及在d位置上的LiB12N12的前线分子轨道Fig.3 The most stable structures of Li adsorption on e and d sites,frontier molecular orbitals of LiB12N12on d site(A)the most stable structure of Li adsorption on e site; (B)the most stable structure of Li adsorption on d site; (C)HOMO orbital;(D)LUMO orbital
氢与BN结构材料的吸附包括了内部和外部两种情形.Sun等18的研究结果表明氢分子比较易于以分子的形式吸附在笼子内部,因此,我们首先考虑了氢分子在笼子内部吸附的情形.氢分子在B12N12内的吸附能计算公式为:
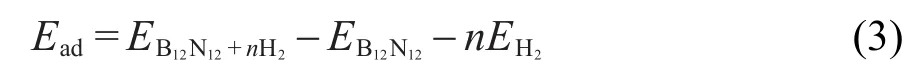
其中,EB12N12+nH2代表了B12N12与H2作用的形成能, EB12N12代表了B12N12的总能量,EH2是单个氢气分子的能量,n是氢气分子的个数.一个氢分子吸附在笼子内部时的形成能是-0.68 eV.当第二个氢分子吸附在里面的时候,形成能迅速减小到-3.10 eV,笼子也向外有所伸展,然后逐渐增加氢气分子个数,当增加到6个时笼子破裂,部分氢以分子形式直接飞出笼子,而有个别的氢分子解离成氢原子吸附在B和N原子的上面,如图4所示.因此,笼子内部最多可以吸附5个氢分子,折合储氢量为3.5%(w).
接下来我们讨论氢分子在LiB12N12笼子外的吸附.当吸附第一个氢分子的时候,发现氢气以分子形式结合在Li原子周围,其吸附能为-0.21 eV.氢分子的键长也被稍微地拉伸到0.076 nm,如图5(a)所示,这个时候Li原子与N的键长也有所拉伸.当吸附两个氢分子时,吸附能为-0.19 eV,分子的键长被拉伸到0.076 nm.这个时候Li原子与N的键长拉伸到0.203 nm,如图5(b)所示.此时,由于极化的作用,氢分子的σ轨道与Li原子周围的前线轨道相互杂化使得氢气以分子的形式与Li成键.通过Bader电荷分析,得到氢分子的两个氢原子与Li原子之间的电荷转移量是不同的,这就是图4(b)中氢分子倾斜地吸附在Li原子周围的原因.为了得到Li原子周围最多能吸附几个氢分子,我们在Li原子的周围加入四个氢分子,不加对称性限制进行几何优化后发现,只有三个氢分子围绕在Li原子周围,而第四个氢分子跑到离Li原子0.421 nm的距离处.因此,最多能有三个氢分子吸附在Li原子周围,如图5(c)所示,氢分子和Li原子距笼子的距离都有所拉伸,氢分子的平均吸附能也继续增加到-0.18 eV.
前面的讨论得到三个Li原子吸附在B12N12笼子周围,Li原子的平均吸附能相对于单个Li原子的吸附能大大减弱.接下来我们在每个Li原子周围都放上三个氢分子,但B12N12笼子仍有两个四元环的位置空闲出来,我们在此地方也加上氢分子.经过充分弛豫之后,三个氢分子仍旧牢固地以化学吸附的形式结合在每个Li原子周围,两个氢分子能够以物理吸附的形式吸附在四元环与六元环相邻的B-N键上,如图5(d)所示.此时每个氢分子与笼子作用的平均结合能是-0.14 eV,折合储氢量为6.5%(w).
以上分别考虑了笼内和笼外的吸附,下面考虑氢气同时吸附在Li修饰的B12N12笼内和笼外的情形.我们优化了氢分子吸附在笼子外部的构型图,如图5(d)所示.结构优化后发现整个体系仍能够保持稳定的构型,对称吸附的Li原子的s轨道与B12N12笼子相互作用使得靠近Li原子周围的氢分子以Li为中心结合在一起.另外的氢分子由于与B12N12笼子的轨道杂化和极化作用而使得氢分子与笼子相互结合在一起.这样共有16个氢分子吸附在Li修饰的B12N12笼子内外,对应的储氢量为9.1%(w).

图4 氢分子吸附在B12N12笼子内部的优化结构Fig.4 Optimized structures of B12N12cage with hydrogen molecule adsorption

图5 氢分子吸附在Li原子修饰的B12N12笼外构型图Fig.5 Optimized structures of Li atom decorated B12N12 cage with hydrogen molecule adsorption (a)Li+1H2;(b)Li+2H2;(c)Li+3H2;(d)3Li+9H2+2H2
4 结论
采用基于密度泛函理论的第一性原理方法研究了Li掺杂的B12N12笼子的储氢行为.研究表明单个Li原子吸附在B12N12笼子的六元环和四元环相交的B-N桥位上相对于其它六个高对称吸附位置更稳定,但是当吸附到三个Li原子的时候发现同时吸附在N的顶位是最稳定吸附位.B12N12笼子的内部最多吸附5个氢分子,储氢量达到3.5%(w);外部最多可以吸附11个氢分子,储氢量达到6.5%(w);考虑到笼内和笼外情形,最大储氢量为9.1%(w).
(1) Golberg,D.;Bando,Y.;Stephan,O.;Kurashima,K.Appl.Phys. Lett.1998,73,2441.doi:10.1063/1.122475
(2) Tang,C.C.;Bando,Y.;Ding,X.X.;Qi,S.;Golberg,D.J.Am. Chem.Soc.2002,124,14550.doi:10.1021/ja028051e
(3) Ma,R.;Bando,Y.;Zhu,H.;Sato,T.;Xu,C.;Wu,D.J.Am. Chem.Soc.2002,124,7672.doi:10.1021/ja026030e
(4) Oku,T.;Kuno,M.Diamond Relat.Mater.2003,12,840.doi: 10.1016/S0925-9635(02)00326-6
(5) Oku,T.;Kuno,M.;Narita,I.J.Phys.Chem.Solids 2004,65, 549.doi:10.1016/j.jpcs.2003.10.033
(6) Narita,I.;Oku,T.Diamond Relat.Mater.2002,11,945.doi: 10.1016/S0925-9635(01)00536-2
(7) Oku,T.;Narita,I.Physica B 2002,323,216.doi:10.1016/ S0921-4526(02)00959-6
(8) Fowler,P.W.;Heine,T.Mitchell,D.;Schmidt,R.;Seifert,G. J.Chem.Soc.Faraday Trans.1996,92,2197.doi:10.1039/ ft9969202197
(9) Chattrarj,P.K.;Bandaru,S.;Mondal,S.J.Phys.Chem.A 2011, 115,187.doi:10.1021/jp109515a
(10)Wen,S.H.;Deng,W.Q.;Han,K.L.J.Phys.Chem.C 2008, 112,12195.
(11) Venkataramanan,N.S.;Note,R.;Sahara,R.;Mizuseki,H.; Kawazoe,Y.Chem.Phys.2010,377,54.doi:10.1016/ j.chemphys.2010.08.015
(12) Shevlin,S.A.;Guo,Z.X.Appl.Phys.Lett.2006,89,153104. doi:10.1063/1.2360232
(13) (a)Yildirim,T.;Ciraci,S.Phys.Rev.Lett.2005,94,175501. (b)Durgun,E.;Ciraci,S.;Zhou,W.;Yildirim,T.Phys.Rev. Lett.2006,97,226102. (c)Durgun,E.;Jang,Y.R.;Ciraci,S.Phys.Rev.B 2007,76, 073413.doi:10.1103/PhysRevB.76.073413
(14) (a)Deng,W.Q.;Xu,X.;Goddard,W.A.Phys.Rev.Lett.2004, 92,166103. (b)Shin,W.H.;Yang,S.H.;Kang,J.K.;Goddard,W.A.Appl. Phys.Lett.2006,88,053111.doi:10.1063/1.2168775
(15) (a)Zhao,Y.F.;Kim,Y.H.;Dillon,A.C.;Heben,M.J.;Zhang, S.B.Phys.Rev.Lett.2005,94,155504. (b)Zhao,Y.F.;Dillon,A.C.;Kim,Y.H.;Heben,M.J.;Zhang, S.B.Chem.Phys.Lett.2006,425,273. (c)Zhao,Y.F.;Lusk,M.T.;Dillon,A.C.;Heben,M.J.;Zhang, S.B.Nano Lett.2008,8,157.doi:10.1021/n1072321f
(16) Sun,O.;Wang,Q.;Jena,P.;Kawazoe,Y.J.Am.Chem.Soc. 2005,127,14582.doi:10.1021/ja0550125
(17) (a)Wu,X.J.;Yang,J.L.;Hou,J.G.;Zhu,Q.S.Phys.Rev.B 2004,69,153411. (b)Wu,X.J.;Yang,J.L.;Zeng,X.C.J.Chem.Phys.2006, 125,044704.
(18) Sun,Q.;Wang,Q.;Jena,P.Nano Lett.2005,5,1273.doi: 10.1021/nl050385p
(19) Beheshtian,J.;Bagheri,Z.;Kamfiroozi,M.;Ahmadi,A.J.Mol. Model.2012,18,2653.doi:10.007/s00894-011-1286-y
(20)Yang,S.;Yoon,M.;Wang,E.;Zhang,Z.J.Chem.Phys.2008, 129,134707.doi:10.1063/1.2981043
February 13,2012;Revised:May 7,2012;Published on Web:May 9,2012.
Density Functional Theory Study on Li-Decorated B12N12Cage for Hydrogen Storage Behavior
XU Wen-Jie HU Zi-Yu SHAO Xiao-Hong*
(College of Science,Beijing University of Chemical Technology,Beijing 100029,P.R.China)
Hydrogen storage behavior in a Li-decorated B12N12cage is investigated using first-principles calculations based on density functional theory(DFT).In the optimized adsorption structure,three Li atoms are adsorbed above the N atom of the B12N12cage(Top-N site).Each Li atom is adsorbed on the bridge site of B-N between the four-and six-membered rings.In addition,each Li atom in the B12N12cage adsorbs three H2molecules,and two H2molecules are adsorbed outside the B12N12cage,with an average H2adsorption energy of-0.14 eV.Inside the B12N12cage,the adsorbed hydrogen remains in the molecular form.Our work shows that the maximum hydrogen storage capacity of Li-decorated B12N12cage is 9.1%(w).
First-principles;Decoration;B12N12;Hydrogen storage;Adsorption energy
10.3866/PKU.WHXB201205091
O641
∗Corresponding author.Email:shaoxh@mail.buct.edu.cn;Tel:+86-10-64433867.
The project was supported by the National Natural Science Foundation of China(51102009)and Fundamental Research Funds for the Central Universities,China(JD1109).
国家自然科学基金(51102009)和中央高校基础基金(JD1109)资助项目
