p38-MAPK信号通路在HHPH中的作用及三七总皂苷干预*
2012-08-30朱阿楠王淑君王园园金可可王万铁
朱阿楠,王淑君,王园园,金可可,王万铁△
(1.温州医学院病理生理学教研室,浙江 温州 325035;2.余姚市中医医院内科,浙江 余姚 315400)
低O2高CO2性肺动脉高压(hypoxic hypercapnia pulmonary hypertension,HHPH)发病的中心环节是肺血管阻力增加。目前研究[1,2]表明HHPH是由于多种因素刺激肺血管平滑肌细胞(vascular smooth muscle cell,VSMC)增殖,细胞外基质成分增多,非肌型动脉形成新肌层化,内皮细胞肿胀和肥大,最终导致肺动脉管壁增厚和管腔狭窄所致。至今,HHPH发生的细胞内信号机制仍未阐明。丝裂原活化蛋白激酶(mitogen activated protein kinase,MAPK)信号途径作为各种刺激因子在细胞内信号传递的共同通路,在肺血管收缩和肺血管结构重建中所起的关键作用,是近年来关注的热点之一。
1 材料与方法
1.1 慢性HHPH动物模型的复制
雄性SD大鼠72只(体重200~220 g),随机分成6 组(n=12),即 N 、H3d、H1w、H2w、H4w和 Hp组 。 将后五组大鼠放入常压低O2高 CO2舱内,舱内O2浓度维持在9%~11%,CO2浓度维持在5%~6%(舱内水蒸汽用无水 CaCl2吸收,多余二氧化碳用氢氧化钠吸收),每天8 h,每周6 d。Hp组于每日进舱前半小时腹腔注射血塞通注射液[50 mg/(kg·d)],其余各组大鼠腹腔注射等量生理盐水。N组置于舱外,自由呼吸空气。其他组均饲养在常压低O2高CO2舱内,分别饲养3 d(H3d组)、1周(H1w组)、2周(H2w组)、4周(H4w、Hp组)。
1.2 模型是否成功的评估
每组随机取10只大鼠。采用右心导管法自颈外静脉插管至肺动脉,并行颈总动脉插管,经Power-Lab生理记录仪分别测定平均肺动脉压(mean pulmonary arterial pressure,mPAP)及平均颈动脉压(mean carotid arterial pressure,mCAP)。放血处死动物,分别称取右室(RV)和左室加室间隔(LV+S)的重量,并计算出RV/(LV+S)的重量比,作为右室肥大的指标。以mPAP、mCAP 、RV/(LV+S)%、作为判断模型建立成功的指标。
1.3 肺细小血管显微结构观察
取肺组织,常规石蜡制片(厚度 5 μ m),HE染色,显微镜观察并拍照。每只大鼠随机选一张肺组织切片,每张切片随机选直径50 μ m~ 200 μ m 肺细小动脉5支,用美国IPP5.0(Image-pro plus)彩色图像分析系统测定肺细小动脉管壁面积/管总面积(vessel wall area/total area,WA/TA),管腔面积/管总面积(vessel cavity area/total area,EA/TA)。
1.4 肺细小动脉p38MAPK的免疫组化检测及结果处理
肺组织石蜡切片常规脱蜡至水,3%过氧化氢室温处理10 min,滴加一抗(1∶100),37℃孵育1 h,再滴二抗、SABC、3,3一二氨基苯联胺(DAB)显色,阳性结果呈棕黄色。随机选取直径50~200 μ m的肺内动脉10条,采用美国IPP5.0(Image-pro plus)彩色图像分析系统测定阳性部位及背景的光密度值,以阳性部位的光密度值减背景的光密度值代表阳性部位的吸光度(optical density,OD)值。
1.5 蛋白免疫印迹法检测肺组织中磷酸化p38MAPK蛋白含量及图像处理
(1)各组取肺组织100 mg,剪碎、匀浆以充分裂解肺组织。超声粉碎仪裂解细胞后吸取上清,分装,-80℃保存。(2)BCA蛋白定量试剂盒检测蛋白浓度:根据标准曲线得出样品的蛋白浓度,调节蛋白浓度,上样量为40 μ g。(3)蛋白质电泳和印迹电转移:SDS-聚丙烯酰胺凝胶电泳配置质量分数为10%的分离胶和5%的浓缩胶,电泳条件:恒压,先80 V,待溴酚蓝到达分离胶后改电压为130 V,直至溴酚蓝跑到凝胶底部;电泳结束后进行印迹转移,应用聚乙烯二氟(PVDF)膜380 mA恒流电转40 min,转移完毕后分别用立春红和考马斯亮兰染膜和胶。(4)Western印迹分析:采用质量分数为5%BSA的TBS。T封闭液室温封闭1 h,TBS-T洗膜3次后,加入单克隆兔抗大鼠磷酸化p38或单克隆兔抗大鼠总p38(总p38为磷酸化p38起校正内参的作用,浓度均为1∶1 000稀释),于4℃孵育过夜,次日加入辣根过氧化物酶标记的羊抗兔IgG(1∶2 000稀释),室温置1 h;TBST洗膜3次,化学发光底物ECL显色、胶片曝光,用UVP凝胶成像扫描系统对胶片条带进行定量性密度测定,Quantity One软件计算其灰度值。
1.6 统计学处理
2 结果
2.1 各组大鼠平均肺动脉压、体循环压和右心指数的比较
2.1.1 各组大鼠 mCAP的比较 N组、H3d组、H1w组、H2w组、H4w组和Hp组大鼠间mCAP无明显差异(P>0.05,表1),说明低O2高CO2对大鼠的体循环压没有影响。
2.1.2 各组大鼠mPAP的比较 与N组相比,H3d组mPAP无明显升高(P>0.05)。随着低O2高CO2时间的延长 ,H1w组 、H2w组、H4w组和Hp组的 mPAP均高于N组(P均<0.05),且各组之间差异明显(P<0.05)。说明随着低O2高CO2时间的延长,大鼠mPAP 1周时即开始明显升高,4周后达到较高水平。同时可见 Hp组 mPAP明显低于H4w组(P<0.05,表1),说明PNS可减轻低O2高CO2的肺动脉高压。
2.1.3 各组大鼠RV/LV+S的比较 随着低O2高CO2时间的延长 ,H2w、H4w和Hp组的 RV/LV+S 均高于N 组(P均 <0.05),但H3d组、H1w组的RV/LV+S较N组增加不明显(P均>0.05)。说明随着低O2高CO2时间的延长,大鼠2周后 RV/LV+S即开始明显升高,而且逐渐增加,第4周达较高水平。同时可见Hp组 RV/LV+S明显低于H4w组(P<0.05,表1),说明PNS可减轻低O2高CO2大鼠的右心室肥厚。
Tab.1 Effect of chronic hypoxia hypercapnia on mPAP,mCAP,RV/LV+S of rats(±s,n=10)

Tab.1 Effect of chronic hypoxia hypercapnia on mPAP,mCAP,RV/LV+S of rats(±s,n=10)
mPAP:Mean pulmonary arterial pressure;mCAP:Mean carotid arterial pressure;R V/LV+S:Right ventricle/left ventricle+septum;N:Normal group;H3d:Hypoxic hypercapnia for 3-day group;H1w:Hypoxic hypercapnia for 1-week group;H2w:Hypoxic hypercapnia for 2-week group;H4w:Hypoxic hypercapnia for 4-week group;HP:PNS-injected group*P <0.05 vs control group;#P <0.05 vs H3dgroup;△P <0.05 vs H1wgroup;▲P<0.05 vs H2wgroup;○P<0.05 vs H4w group
Group mPAP(mmHg) mCAP(mmHg)RV/LV+S(%)N 11.00±0.62 107.46±6.40 23.41±0.97 H3d 11.84±0.49 110.13±5.44 24.29±1.02 H1w 13.54±0.65*# 109.19±4.99 24.76±1.04 H2w 16.85±0.88*#△ 108.03±4.9230.19±0.95*H4w 23.12±0.53*#△▲ 114.85±9.8736.04±0.65*#△Hp 18.74±0.74*#△▲○106.32±6.3431.57±0.69*#△○
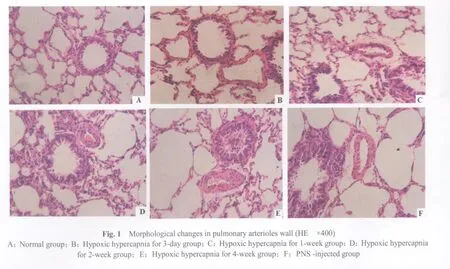
2.2 光镜下各组大鼠肺细小动脉结构的比较
光镜下所见,N组,平滑肌层未见明显增生,管壁均匀一致,而随着缺氧间期的延长,H1w组、H2w组、H4w组和Hp组内弹力板扭曲明显,中膜平滑肌细胞增生,管腔明显狭窄,但H3d组改变不明显(图1A,1F,图1见彩图页Ⅳ)。Hp组与H4w组比较,其血管壁的改变明显减轻(图1E,1F)。随着低O2高CO2时间的延长,H1w、H2w、H4w和Hp组的WA/TA 均高于N组(P均<0.05),但H3d组较N组增加不明显(P>0.05)。说明随着低氧时间的延长,大鼠1周后肺小动脉管壁开始增厚,第4周达较高水平。同时可见Hp组WA/TA明显低于H4w组(P<0.05),说明PNS可减轻低O2高CO2大鼠肺小动脉管壁增厚。EA/TA则相反(表2)。
Tab.2 Effect of chronic hypoxia hypercapnia on pulmonary arteriolar remodeling of rats(%,±s,n=8)
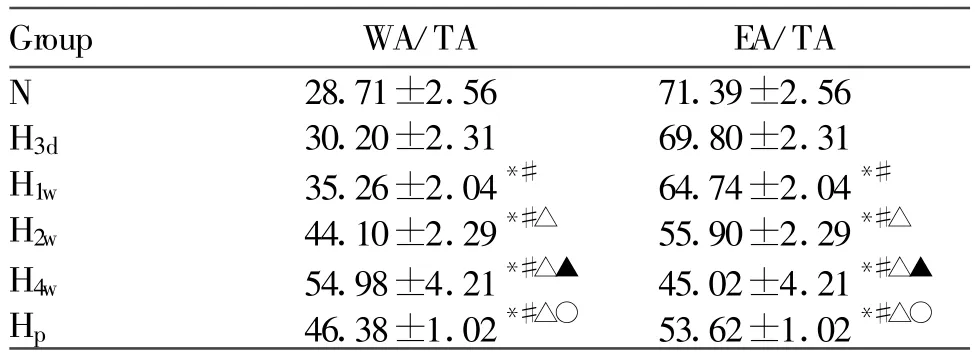
Tab.2 Effect of chronic hypoxia hypercapnia on pulmonary arteriolar remodeling of rats(%,±s,n=8)
WA/TA:Vessel wall area/total area;EA/TA:Vessel cavity area/total area;N:Normal group;H3d:Hypoxic hypercapnia for 3-day group;H1w:Hypoxic hypercapnia for 1-week group;H2w:Hypoxic hypercapnia for 2-week group;H4w:Hypoxic hypercapnia for 4-week group;HP:PNS-injected group*P<0.05 vs control group;#P<0.05 vs H3dgroup;△P<0.05 vs H1wgroup;▲P<0.05 vs H2wgroup;○P<0.05 vs H4w group
Group WA/TA EA/TA N 28.71±2.56 71.39±2.56 H3d 30.20±2.31 69.80±2.31 H1w 35.26±2.04*# 64.74±2.04*#H2w 44.10±2.29*#△ 55.90±2.29*#△H4w 54.98±4.21*#△▲ 45.02±4.21*#△▲Hp 46.38±1.02*#△○ 53.62±1.02*#△○
2.3 各组肺组织匀浆磷酸化p38MAPK(P-p38MAPK)Western blot结果比较
各组肺组织匀浆P-p38MAPK灰度值变化为N组P-p38 MAPK未见表达,随着低O2高CO2进展,p38MAPK活性逐渐增强,H1w组达到高峰(图2),灰度值(0.362±0.087),此后一直维持在较高水平,H2w、H4w和H1w组之间无统计学差异。Hp组 P-p38 MAPK明显低于H1w、H2w、H4w组但仍高于正常对照组水平(P<0.05,表3)。

Fig.2 Western blot analysis of P-p38MAPK expression in lung homogenate
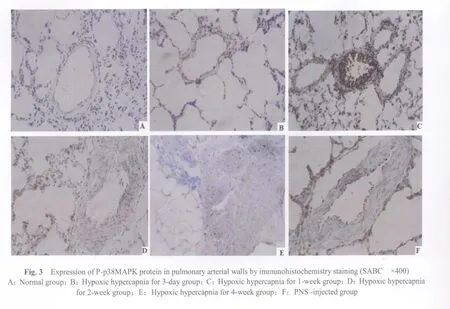
2.4 各组肺小血管壁磷酸化p38MAPK蛋白表达变化
各组肺小动脉壁 P-p38MAPK蛋白吸光度值比较N组和H3d组肺小动脉壁P-p38MAPK表达呈阴性或弱阳性(图3A、3B,图3见彩图页Ⅳ),随着低O2高CO2进展,其在肺小动脉壁上的表达逐渐增强(表4),低氧1周时P-p38MAPK在大鼠肺小动脉壁核表达呈阳性(图3C),低O2高CO2两周,4周时也有较强活性(图3D,3E)。PNS治疗组P-p38MAPK表达减弱(图3F,表3)
Tab.3 Western blot analysis of P-p38MAPK expression in lung homogenate(±s,n=6)and expression of P-p38MAPK protein in pulmonary arterial walls(±s,n=10)
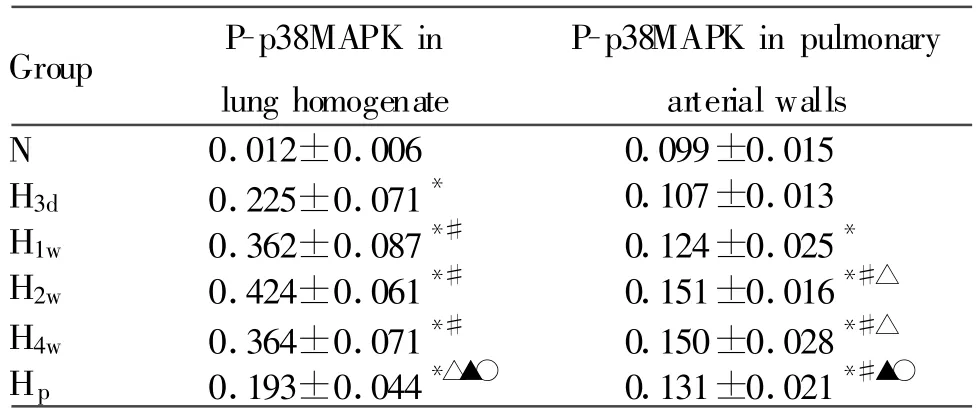
Tab.3 Western blot analysis of P-p38MAPK expression in lung homogenate(±s,n=6)and expression of P-p38MAPK protein in pulmonary arterial walls(±s,n=10)
N:Normal group;H3d:Hypoxic hypercapnia for 3-day group;H1w:Hypoxic hypercapnia for 1-week group;H2w:Hypoxic hypercapnia for 2-week group;H4w:Hypoxic hypercapnia for 4-week group;HP:PNS-injected group*P<0.05 vs control group;#P<0.05 vs H3dgroup;△P<0.05 vs H1wgroup;▲P<0.05 vs H2wgroup;○P<0.05 vs H4w group
Group P-p38MAPK in lung homogenate P-p38MAPK in pulmonary arterial walls N 0.012±0.006 0.099±0.015 H3d 0.225±0.071* 0.107±0.013 H1w 0.362±0.087*# 0.124±0.025*H2w 0.424±0.061*# 0.151±0.016*#△H4w 0.364±0.071*# 0.150±0.028*#△Hp 0.193±0.044*△▲○ 0.131±0.021*#▲○
3 讨论
本实验结果表明,随着低O2高CO2时间的延长和模型复制的进展,大鼠肺动脉平均压和右心指数呈动态增高,两周组和4周组均显著高于正常对照组,光镜下显示肺细小动脉内弹力板扭曲,中层平滑肌及外膜胶原纤维增生,管腔狭窄,说明低O2高CO2肺动脉高压和右心室肥厚模型已建立成功。肺血管重建不但是肺动脉高压持续发展的关键因素,而且是对血管扩张降压药物产生抵抗的主要原因。目前认为,在低O2、高碳酸等各种刺激因子作用下,肺动脉平滑肌细胞增殖以及胶原等细胞外基质在管壁大量沉积是肺动脉重建的最主要的原因。我们的实验发现:随着低O2高CO2时间的延长,(1)大鼠mPAP 1周后即开始明显升高,4周时达到高峰。(2)2周组RV/LV+S即开始明显升高,4周达最高水平。(3)2周组、4周组内弹力板扭曲明显,中膜平滑肌细胞增生,管腔明显狭窄。说明在HHPH形成过程中出现明显的肺血管重建。
Kato等[3]研究表明血小板源生长因子(platelet derived growth factor,PDGF)对新生内膜的VSMC和成熟的中膜VSMC的MAPK激活有相同的作用。Su[4]等发现 ROS通过p38MAPK信号通路促进了VSMC分化。Viedt等[5]观察到 ROS调节了AngⅡ诱导的JNK和p38MAPK激活,使大鼠VSMCs增殖加快,认为表型转化可能与ROS激活了MAPK通路有关。Kavurma等[6]研究证明不同细胞亚型的增殖和迁移是MAPK依赖性的,并证明SMC异质性至少部分地被特定的MAPK通路所调节。我们的实验也发现,(1)肺组织P-p38MAPK水平在1周时即达到峰值,且此后一直维持在峰值水平。(2)肺小动脉壁P-p38MAPK蛋白吸光度值变化基本同肺组织匀浆Western blot结果一致;且随着低O2高CO2进展,其主要表达在肺小动脉中膜和内膜,外膜也有少量表达。这些结果表明低O2高CO2能够引起p38MAPK通路的活化;P-p38MAPK在HHPH中呈动态变化;它们的活化参与了肺动脉高压的形成以及肺血管的重建。但我们的实验结果与文献报道并不完全一致:孔春初[7]等采用单纯低O2复制大鼠肺动脉高压,P-p38MAPK在正常组和各低氧组肺组织均未见表达,造成实验结果差异的机理尚不清楚,有可能是高碳酸血症增强P38 MAPK活化。值得一提的是,P-p38MAPK蛋白在低O2高CO23d时活性增强,在1周达到高峰后一直保持在高活化状态。说明低O2高CO2中晚期参与血管重塑的主要通路可能是p38MAPK。Nagata研究[8]发现,p38 MAPK促进凋亡或抑制凋亡与p38 MAPK激活的性质有关,如短期p38MAPK的激活可引起造血肿瘤细胞KT6分化,而长时间的激活则可促进其凋亡;在TNF-α处理的中性粒细胞中早期p38的激活可延迟凋亡,而晚期p38的激活可加速凋亡。因此,也可以认为中晚期p38 MAPK高活化状态是一种保护机制,以此来对抗低O2高CO2引起的肺血管壁增殖重塑。p38 MAPK在HHPH中晚期的作用机制有待于进一步研究。
三七总皂苷(panax notogino side,PNS)具有扩张血管、降低血液粘度等多种作用 。我们的实验结果显示,PNS用药组大鼠与慢性HHPH模型组大鼠相比,平均肺动脉压和右心室重量比都有不同程度的降低,而颈总动脉压无明显改变,提示PNS可抑制或预防慢性HHPH的形成而对体循环压无明显影响,即对HHPH具有选择性抑制作用。光镜下Hp组肺细小动脉内弹力板扭曲明显减轻,中层平滑肌变薄,WA/TA明显降低,LA/TA显著升高。提示PNS具有抑制慢性HHPH大鼠的肺血管结构重建作用。以往研究表明[9],PNS可通过增加血浆、肺组织中NOS活性和NO含量,增加血浆中T-SOD、Cu/ZnSOD活性,降低红细胞比积和减轻内皮细胞损伤而实现防治慢性低氧性肺动脉高压的形成。周晓霞[10]研究提示 PNS的抗动脉硬化作用可能是通过抑制VSMC异常增殖实现的。我们的实验表明,Hp组大鼠肺组织,肺动脉壁上的P-p38MAPK蛋白表达均有下降,同时Hp组肺血管结构重建较4周组 显著减轻,说明PNS能够抑制p38MAPK通路活性,从而减轻肺动脉高压的程度。
[1]Fagan K A,Oka M,Bauer N R,et al.Attenuation of acute hypoxic pulmonary vasoconstriction and hypoxic pulmonary hypertension in mice by inhibition of Rho-kinase[J].Am J Physiol Lung Cell Mol Physiol,2004,287(4):L656-664.
[2]Nagaoka T,Morio Y,Casanova N,et al.Rho/Rho-kinase signaling mediates increased basal pulmonary vascular tone in chronically hypoxic rats[J].Am J Physiol Lung Cell Mol Physio l,2004,287(4):L665-672.
[3]Kato M,Tanimoto A,Arima N,et al.Response to plateletderived growth factor by phenotypically different cultured human aortic smooth muscle cells[J].Biochem Mol Biol Int,1998,44(4):815-823.
[4]Su B,Mitra S,Gregg H,et al.Redox regulation of vascular smooth muscle cell differentiation[J].Circ Res,2001,89(1):39-46.
[5]Viedt C,Soto U,Krieger-Brauer H I,et al.Differential activation of mitogen-activated protein kinases in smooth muscle cells by angiotensin II:involvement of p22phox and reactive oxygen species[J].Arterioscler Thromb Vasc Biol,2000,20(4):940-948.
[6]Kavurma M M,Khachigian L M.ERK,JNK,and p38MAP kinases differentially regulate proliferation and migration of phenotypically distinct smooth muscle cell subtypes[J].J Cell Biochem,2003,89(2):289-300.
[7]孔春初,戴爱国.丝裂原活化蛋白激酶调节缺氧诱导因子1α对大鼠缺氧性肺动脉高压的作用[J].中华结核和呼吸杂志,2005,28(5):328-332.
[8]Nagata Y,Todokoro K.Requirement of activation of JNK and p38 for environmental stress-induced erythroid differentiation and apoptosis and inhibition of ERK for apoptosis[J].Blood,1999,94(3):853-863.
[9]周晓霞,苏佩清,杨鹤梅.三七总皂甙对人高脂血清诱发的胎儿血管平滑肌细胞增殖的抑制作用[J].中国动脉硬化杂志,2000,8(1):43-45.
[10]马文裴,杨映宁,周定邦,等.三七总皂甙对慢性低氧性肺动脉高压大鼠的防治作用[J].中国病理生理杂,2004,20(6):1074-1077.
