海洋沉积物的铁和锌同位素测定
2012-08-03祁昌实朱祥坤戴民汉唐索寒李志红李世珍
祁昌实, 朱祥坤, 戴民汉, 唐索寒, 吴 曼,李志红, 李世珍, 李 津
(1.厦门大学 近海海洋环境科学国家重点实验室, 福建 厦门 361005; 2.中国地质科学院地质研究所 国土资源部同位素地质重点实验室, 北京 100037; 3.云南大学 云南省古生物研究重点实验室, 云南 昆明 650091)
0 引 言
Fe和Zn是海洋浮游植物生长和新陈代谢所必需的痕量营养元素[1–2]。有学者提出“Fe假说”[3]和“Zn假说”[4], 认为部分海域表层海水Fe或无机Zn是海洋初级生产力的限制因子。最近的研究显示,陆架沉积物可能是海水Fe的重要源区[5]。然而, 受采样、样品制备和分析技术的限制[6], 研究沉积物-海水间的Fe和Zn的循环过程仍存在较大困难。
近十年来, 基于多接收器电感耦合等离子体质谱(MC-ICPMS)测试技术研究Fe和Zn同位素地球化学取得了快速发展[7–15]。目前的同位素分馏实验研究显示, 溶解或沉淀、生物作用、氧化还原和吸附等过程可导致 Fe[16–20]或 Zn[21–24]同位素的质量分馏,因此Fe同位素和Zn同位素可能是一种示踪海洋Fe和Zn的生物地球化学循环过程的重要手段。在波罗的海全新世沉积物中, Fehret al.[25]发现, 相对于氧化底层水环境下沉积的沉积物, 静海相沉积物有明显偏轻的 Fe同位素信号; 在黑海, Severmannet al.[26]发现, 相比静海相沉积物, 氧化大陆架沉积物有明显偏重的Fe同位素信号。上述Fe同位素信号差异可用“有轻同位素信号的 Fe2+从浅部大陆架沉积物向深静水盆地输出”的模型解释[25–26], 这暗示沉积物Fe同位素对示踪大陆架沉积物-海水间的Fe循环过程具有重要意义。但截止目前, 总体来说, 报道的海洋沉积物Fe和Zn同位素数据仍然十分有限。
相对于岩石样品, 海洋沉积物通常具有高的含盐量, 部分高生产力海区的沉积物还具有富含有机质的特征。实验表明, 有机物质和 Al等基质与 Fe或Zn的不完全化学分离将产生“基质效应”(matrix effects), 导致样品标准交叉法(sample-standard bracketing)进行仪器质量分馏校正后的同位素数据偏离真值[7,14,27]。另外, 有机物质可能络合溶液中的Fe和 Zn离子; 在随后的离子交换柱分离纯化过程中, 该部分的 Fe和 Zn可能表现出与 Fe3+和Zn2+无机离子不一致的行为, 从而无法有效回收, 并导致相关的二次同位素分馏。因此, 基于常规地质样品建立的Fe、Zn同位素分析的样品前处理流程不能完全适用于海洋沉积物的Fe和Zn同位素分析, 建立针对海洋沉积物的Fe和Zn同位素分析方法具有重要意义。目前的海洋沉积物Fe和Zn同位素样品制备流程中的海盐和有机物质处理主要依据文献[7]的方法: 将样品置于纤维素微孔滤膜上, 用去离子水淋洗海盐, 减少Na+、Mg2+和Al3+等基体元素阳离子以及Cl−等阴离子数量, 并避免NaCl在上柱HCl中饱和析出; 使用浓 HNO3消解样品中的有机物质和滤膜。该前处理方法繁琐耗时, 过滤的真空泵不适合在洁净实验室内进行使用; 过滤步骤还可能改变海洋沉积物的pH值, 引起处于吸附态的Zn离子的丢失[28–29]和相关的二次同位素分馏[24]。另外, 在消解海洋沉积物内部大量种类复杂的有机物质时, 浓HNO3常常无法将有机物完全转化为无机组分。针对以上问题, 我们研究建立了一种新的适用于海洋沉积物Fe和Zn同位素同位素分析的化学前处理方法,并对南海西部夏季上升流区两个沉积物柱样的 Fe和Zn同位素组成进行了测定。
1 样 品
沉积物样品 Y32柱样和 Y20柱样分别采集于2006年 12月和 2007年 8~9月, 采样位置分别为13°28.55′N、110°11.14′E 和 11°29.68′N、110°02.54′E,采样站位的水深分别为2390 m 和1018 m 。航次由厦门大学“海洋生物地球化学过程和机制”创新研究群体组织。两采样站位均位于南海西部夏季上升流区(图1)。海洋沉积物用箱式采泥器进行采集, 现场采用聚丙烯柱套取直径为 5 cm的柱状样。其中,Y32柱样长18 cm, Y20柱样长8 cm(相对于同时采集的最长中心柱样, 该柱样表层1 cm被侵蚀)。柱样立即在现场进行分割(Y32柱样为2 cm/层; Y20柱样为1 cm/层), 样品立刻装入双层干净塑料袋中, 于−18 ℃冷冻保存。分取过程中, 避免样品与金属接触。
Y32柱样和 Y20柱样主要由碳酸钙和粘土组成。Y20柱样有机质含量平均为 1.03%[30]。依据文献[31]对该上升流区 MD05-2901沉积柱样(14°22.50′N、110°44.60′E)的研究(图 1), 该区域的沉积速率估计约为10 cm/ka。
2 分析方法
2.1 样品消解、脱盐及Fe和Zn的分离纯化
样品的消解、脱盐、Fe和Zn的分离纯化均在中国地质科学院国土资源部同位素地质重点实验室内完成。HCl、HNO3和 HF均为优级纯并经二次蒸馏纯化, HClO4为Aldich 超纯试剂(99.999 %), H2O2为北京化学试剂研究所制备的超纯试剂。
沉积物样品经过冷冻干燥后, 用玛瑙钵研磨至200目。取约0.10 g粉末样品, 置于7 mL Savillex Teflon消解罐中, 依次加入1 mL HNO3、2 mL HF和0.5 mL HClO4, 于 120 ℃保温 24 h后, 在 150 ℃下蒸干; 随后, 加入 1 mL 浓 HCl + 30 μL 30% H2O2, 于80 ℃保温24 h后, 蒸干溶液。此时, 有机质将被完全氧化; 加入0.2 mL浓HCl溶解样品, 将清液移出至干净Teflon杯中。加入少量稀盐酸完全溶解残留的结晶盐, 110 ℃蒸发至有晶体开始析出, 加入 0.1 mL浓 HCl; 常温保存片刻后, 将清液吸出, 与前一次转移的清液合并(定义为一遍脱盐)。除 CAGSR-a和 CAGSR-a∗(定义见下文)以外, 其余样品重复一遍脱盐步骤。蒸干合并的清液, 加入 0.6 mL 7 mol/L HCl + 0.001% H2O2完全溶解样品。取适量溶液, 采用文献[32, 33]流程进行 Fe和 Zn的纯化。蒸干 Fe和Zn接收液, 分别转化为HNO3和HCl介质。最后Fe和Zn分别采用0.1 mol/L HNO3和0.1 mol/L HCl溶解[13–14], 进行同位素比值的质谱测定。
为检验实验相关步骤对海洋沉积物Fe和Zn同位素组成的影响, 称取4份玄武岩标准CAGSR-1(每份约0.05 g), 加入约0.02 g NaCl(国药集团生产, 光谱纯), 命名为 CAGSR-a、CAGSR-a∗、CAGSR-b和CAGSR-b∗, 与海洋沉积物样品一同进行消解、脱盐、分离纯化和质谱测定。加入NaCl总量按0.1 g干沉积物原先有0.7 g间隙海水且海水中海盐含量为35‰进行估计。NaCl中的Fe和Zn含量在中国科学院地质与地球物理研究所岩石圈演化国家重点实验室 Agilent 7500 ICP-MS上采用标准加入法测定,NaCl中 Fe 的含量为 8.6 μg/g, Zn 为 0.60 μg/g。
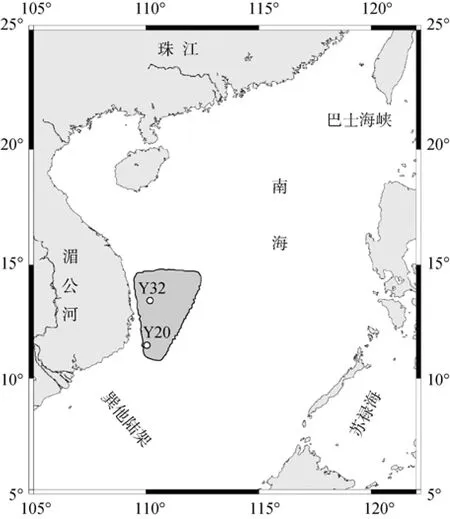
图1 Y32柱样和Y20柱样采样站位图(深灰色阴影部分代表上升流区)Fig.1 Map of South China Sea showing the sampling site.Note that the shadow area is the upwelling region
2.2 沉积物Fe和Zn的同位素比值测定
在国土资源部同位素地质重点实验室的Nu HR MC-ICPMS上完成Fe和Zn同位素的测定。Fe同位素比值测定在高分辨模式下进行。在高分辨模式下,该仪器可以将载气 Ar形成的多原子离子团干扰信号(40Ar14N和40Ar16O)与样品的54Fe和56Fe信号分开,从而获得样品真正的Fe信号。样品溶液与校正标准溶液的Fe浓度匹配至偏差小于10%后, 样品溶液通过DSN-100膜去溶进入等离子体, 在7000 K的高温下等离子化, 然后经电场和磁场双聚焦后, 进入法拉第接收器进行测定。数据采用基于Unix操作系统的控制软件进行自动采集。测定过程中的仪器质量分馏采用样品-标准交叉法进行校正。Fe同位素的分析结果采用相对于国际标准物质 IRMM-014的56Fe/54Fe千分偏差δ56Fe来表示。关于仪器物理参数设定、数据采集和校正方法的详细信息, 参看文献[14]。Zn同位素比值测定在低分辨模式下进行。样品溶液浓度与校正标准溶液 Zn浓度匹配至偏差小于30%后, 样品溶液通过DSN-100膜去溶进入等离子体。测定过程中的仪器质量分馏采用样品-标准交叉法进行校正。对于Zn同位素的分析结果, 除加入NaCl的CAGSR和间隔插入的CAGS-Zn(相对于国际标准物质Romil-Zn)外, 海洋沉积物样品均相对于国际标准物质JMC3-0749C的66Zn/64Zn的千分偏差δ66Zn来表示。测定方法的详细信息, 参看文献[13]。
3 结果与讨论
相比文献[7]采用浓HNO3, HClO4可更快捷地消解有机质, 将其转化为无机组分, 显著减少了样品消解时间。另外, 相比文献[7]流程, 本文脱盐方法无需过滤, 避免了吸附态Zn离子的解吸及其可能带来的丢失和同位素分馏。由于本实验方法的基础是岩石样品Fe和Zn分离纯化流程[32–33], 因此有必要检验: (1)分离纯化后的Fe和Zn组分中是否残留有海洋沉积物特有的基质元素或Fe、Zn的同质异位素等干扰元素; (2)HClO4的使用和本流程中的脱盐方法对Fe和Zn同位素组成影响。
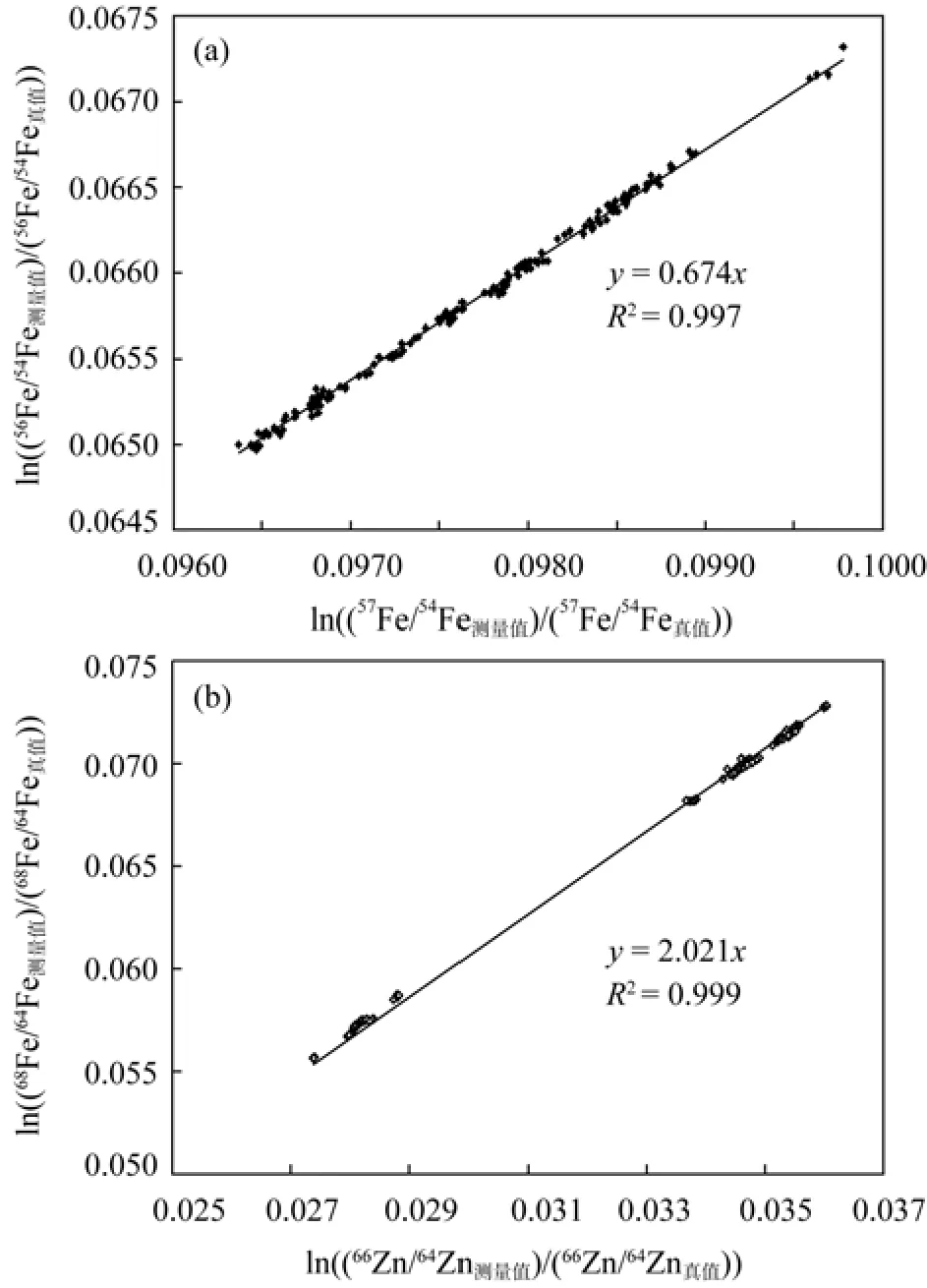
图2 Fe同位素比值数据(a)和Zn同位素比值数据(b)相互关系Fig.2 Three-isotope representation of the natural logarithm of the measured isotope ratios normalized to the true ratios for Fe (a) and Zn (b)
3.1 干扰元素
与岩石样品相比, 海洋沉积物中通常含有大量的Na、Mg、Al和Ca等基质元素。已有研究表明: 当溶液中Al/Fe > 2%(质量比, 下同)时, Al将会对Fe同位素测定产生影响[14]。另外, Al、Ca和Mg可与其他元素一起对Fe或Zn同位素构成同质异位素干扰[13,14,34]。对Fe各同位素存在干扰的同质异位素还包括54Cr、58Ni, Pd、Cd和Sn等相关同位素形成的二价离子以及40Ar形成的双原子离子或多原子离子;对64Zn、66Zn和67Zn存在干扰的同质异位素则包括64Ni, Ba相关同位素形成的二价离子, 以及Ti、Ca、V和Cr与其他原子形成的双原子离子或多原子离子[35]。
在MC-ICPMS上的扫描结果显示, 分离纯化后的 Fe溶液中, 残留 Al/Fe < 0.05%, 残留 Na/Fe、Mg/Fe < 0.01%, 其余干扰元素检测不到。此外, 所有 Fe同位素比值原始数据之间表现出理想的线性关系, 与同位素分馏理论值几乎一致(图2a)。这表明来自于样品和载气的基体元素干扰和同质异位素干扰均得到了很好的控制, 不会对56Fe/54Fe比值和57Fe/54Fe比值产生影响。对分离纯化后的Zn溶液的MC-ICPMS扫描结果显示: 残留 Na/Zn、Mg/Zn和Al/Zn < 0.5%, Ca/Zn、Ti/Zn 和 Cr/Zn < 0.3%, 其余干扰元素检测不到。此外, 所有Zn同位素比值原始数据之间表现出理想的线性关系(图 2b)。这表明影响Zn同位素比值测定的相关干扰均得到很好地去除,不会对66Zn/64Zn比值和67Zn/64Zn比值产生影响。
3.2 高氯酸及脱盐过程
表1显示, 经过含 HClO4混合酸消解和一遍脱盐步骤处理后, 加入 NaCl的两个玄武岩标准CAGSR-1(CAGSR-a 和 CAGSR-a∗)的δ56Fe 值(相对于 IRMM-014)平均值为 0.18‰±0.03‰(2SE),δ66Zn 值平均值(相对于 Romil-Zn)为 7.36‰±0.09‰(2SE); 经过两遍脱盐步骤处理后, 加入 NaCl的两个玄武岩标准 CAGSR-1(CAGSR-b和CAGSR-b∗)的δ56Fe 值(相对于 IRMM-014)平均值为0.19‰±0.06‰(2SE),δ66Zn 值平均值(相对于Romil-Zn)为 7.36‰±0.09‰(2SE)。这与 CAGSR-1的δ56Fe和δ66Zn推荐值[37]在误差范围内一致。经过含HClO4混合酸消解和两次脱盐步骤处理过的海洋沉积物中, Y32-2和Y20-2样品的δ56Fe(相对于IRMM-014)和δ66Zn值(相对于JMC 3-0749L)与平行样亦显示很好的一致性(表2)。上述测定结果表明, 高氯酸的使用以及脱盐步骤没有导致Fe和Zn同位素分馏。
3.3 分析精度评估
样品同位素分析精度的评估通过四个步骤进行。(1)标准溶液的同位素比值长期重现性。对于Fe同位素, 高分辨模式下 CAGS-Fe3溶液在国土资源部同位素地质重点实验室内为期 16个月的测定结果为:δ56Fe=0.83‰±0.08 ‰(2SD, 相对于 IRMM-014)[14]; (2)本次测定过程间隔插入的标准溶液的δ56Fe值重现性。相对于 IRMM-014, 本次试验的CAGS-Fe3溶液的δ56Fe平均值为 0.79‰±0.10‰(2SD,n=13)(图3a); (3)与样品平行操作的、加入NaCl的 CAGSR玄武岩标准的δ56Fe值重现性。相对于IRMM-014, 经过两次脱盐的两个 CAGSR(CAGSR-b和 CAGSR-b∗)的δ56Fe 值差别为 0.05‰(表 1); (4)海洋沉积物样品全流程平行样的δ56Fe值重现性。Y32-2两个平行样的δ56Fe值差别为 0.03‰, Y20-2两个平行样的δ56Fe值差别为 0.02‰(表2)。虽然海洋沉积物平行样的δ56Fe值差别小于实验室标准溶液的分析精度, 也小于加入 NaCl的玄武岩标准的δ56Fe值差别, 考虑到前者统计次数有限且样品纯化溶液与标准金属溶液存在一定差别, 我们选取以上四个步骤中最差精度 0.10‰(2SD)作为本次测试的海洋沉积物样品δ56Fe值外部精度。
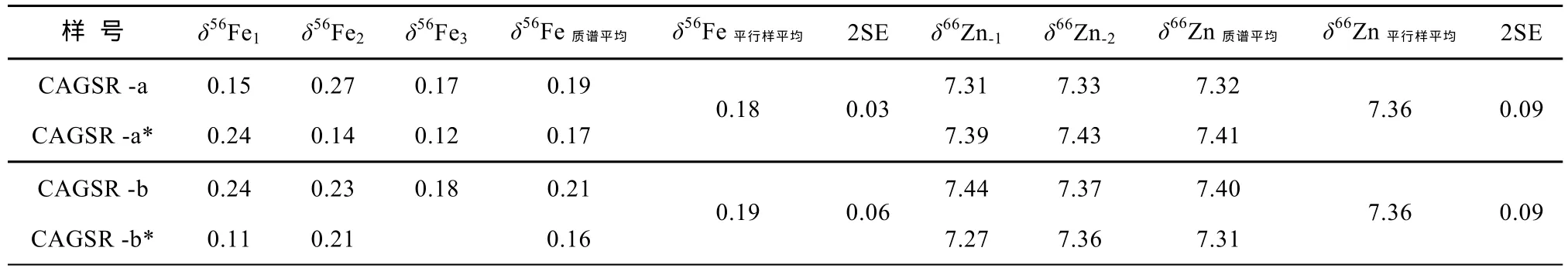
表1 加入NaCl的CAGSR-1的Fe和Zn同位素测定结果Table 1 Results of Fe and Zn isotopic ratios for CAGSR-1 added with NaCl
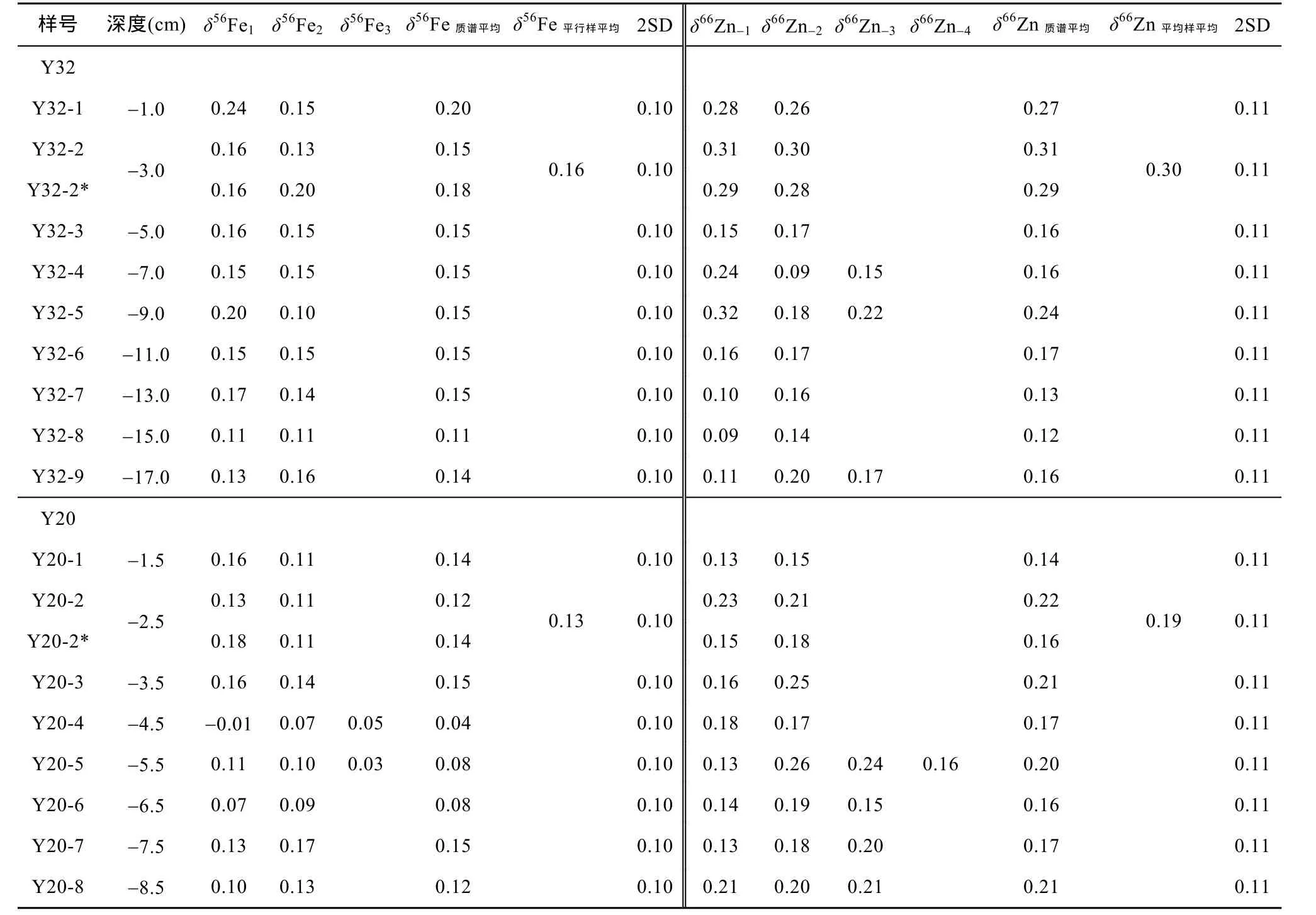
表2 南海西部上升流区柱状沉积物的Fe和Zn同位素组成Table 2 Isotope composition of Fe and Zn of marine sediments in the upwelling region of west of the South China Sea
相比本次实验中加入NaCl的玄武岩标样、海洋沉积物平行样和间隔插入的 CAGS-Zn标准溶液测试精度(图3b), 实验室标准溶液为期7个月的δ66Zn值分析精度 0.11‰[13]略差, 被作为本次实验的海洋沉积物样品δ66Zn值外部精度。
相比目前国际上报道的δ56Fe和δ66Zn分析精度0.10‰(2SD)[7,9,12–14], 本次实验的海洋沉积物样品亦获得了相同或相近的精度。另外, 实验室 USGS玄武岩标样BCR-2的δ56Fe值为0.071‰±0.064‰(2SD,n= 16)[38],δ66Zn值为−0.072‰±0.020 ‰(2SD,n= 19,相对于IRMM Zn)(实验室未发表资料), 与当前国际上其他实验室报道值一致[15,39–41]; 此次实验中加入NaCl的玄武岩标准CAGSR的δ56Fe和δ66Zn值在分析精度内与推荐值一致[37]。因此, 该前处理和Fe、Zn同位素测定方法可获得精确的海洋沉积物Fe、Zn同位素组成。
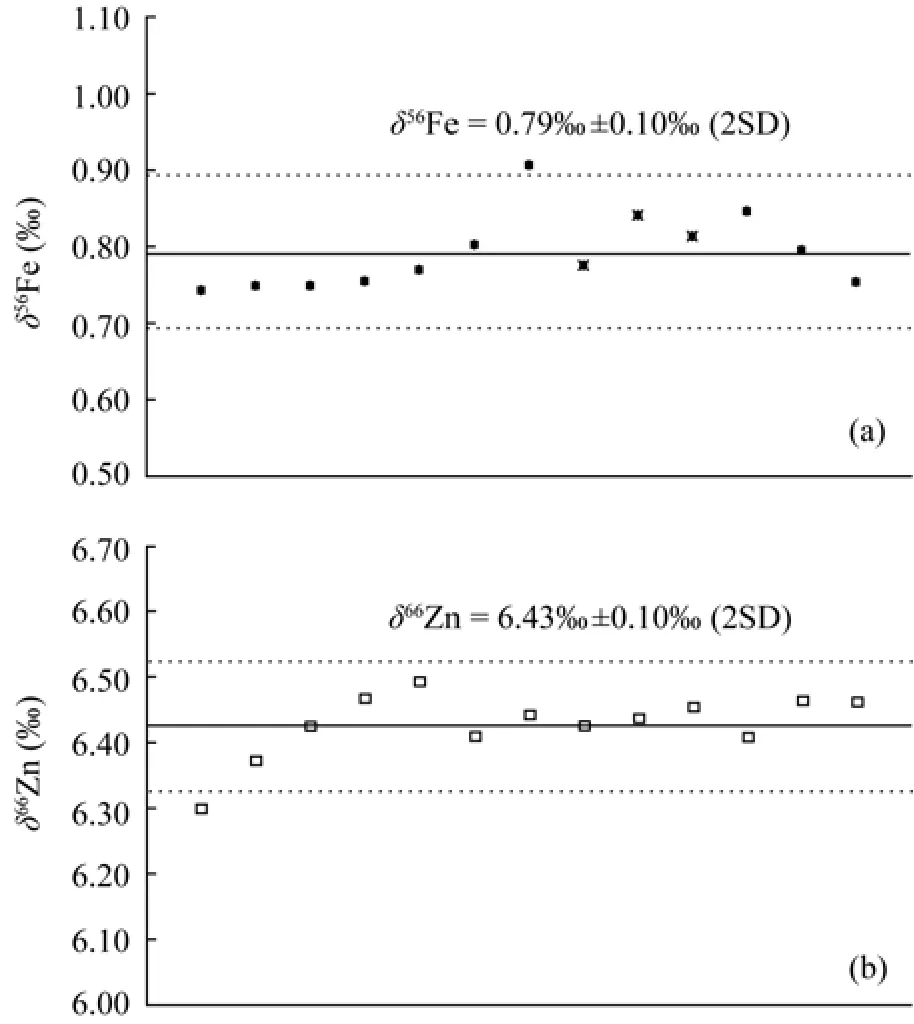
图3 测定过程间隔插入的CAGS-Fe3标准溶液的δ56Fe(相对于IRMM-014)(a)和CAGS-Zn标准溶液的δ66Zn(相对于 Romil-Zn)(b)测定值Fig.3 δ56Fe (3a, relative to IRMM-014) and δ66Zn (3b, relative to Romil-Zn) of inserted internal standard CAGS-Fe3 during determination of marine sediments in this study
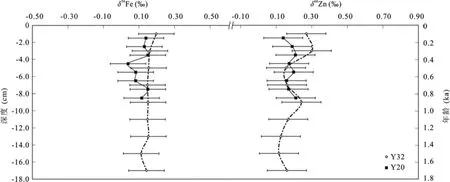
图4 Y32和Y20柱样的Fe同位素和Zn同位素随深度的变化Fig.4 Downcore variations of Fe and Zn isotope compositions at sites Y32 and Y20 in the western South China Sea
3.4 南海西部柱状表层沉积物Fe和Zn同位素组成
表2显示, Y32柱样中, 表层至18 cm柱底部共9件样品的δ56Fe分布范围为 0.11‰~0.20‰, 平均值为 0.15‰±0.04‰(2SD);δ66Zn 分布范围为 0.12‰~0.30‰, 平均值为 0.19‰±0.13‰(2SD)。Y20 柱样中, 从2~9 cm底部共8件样品的δ56Fe分布范围为0.04‰~0.15‰, 平均值为 0.11‰±0.08‰(2SD);δ66Zn分布范围为0.14‰~0.21‰, 平均值为0.18‰±0.05‰(2SD)。总的来说, 不论是 Y32还是 Y20柱,δ56Fe和δ66Zn值随深度均无明显变化; 两个柱之间也没有明显差异(图4)。在下面的讨论中, 将把Y32和Y20柱沉积物作为一个整体, 代表南海西部上升流区约1~2 ka以来的沉积物(简称“南海西部上升流区沉积物”), 与目前国际上报道的海洋样品(包括沉积物和/悬浮颗粒物)、大气沉降样品以及黄土等进行δ56Fe和δ66Zn值对比。
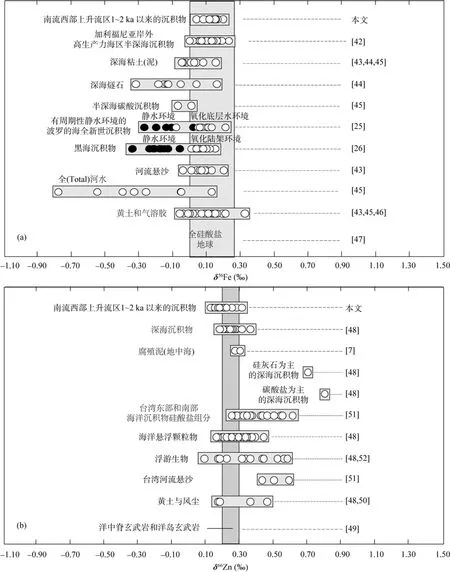
图5 南海西部上升流区1~2 ka沉积物与相关海洋样品以及陆源输出端元的δ56Fe (a)和δ66Zn (b)对比Fig.5 Comparison of isotopic compositions between marine sediments with an age of 1–2 ka in the western South China Sea and marine samples as well as terrestrial sources for δ56Fe (a) and δ66Zn (b)
南海西部上升流区沉积物的δ56Fe范围在0.04‰~0.20‰之间, 与目前报道的加利福尼亚岸外高生产力海区半深海沉积物(0.00‰~0.26 ‰)[42]、河流悬浮颗粒(−0.04‰~0.20‰)[43]和深海粘土[43–45]一致, 也与黄土、气溶胶(−0.06‰~0.33‰)[43,45,46]和火成岩的δ56Fe值[43]一致(图5a), 但略高于深海燧石的δ56Fe值(−0.32‰~0.16‰)[44]。相对于黑海静海相沉积物(−0.34‰~−0.06‰)[26]和波罗的海全新世静海相沉积物(−0.27‰~−0.02‰)[25], 南海西部上升流区沉积物的δ56Fe值明显偏高, 并与黑海氧化陆架(oxic shelf)及波罗的海全新世氧化底层水环境沉积物的δ56Fe值一致(图5)。
南海西部上升流区沉积物的δ66Zn范围为0.12‰~0.30‰, 与目前报道的深海沉积物的δ66Zn范围(0.17‰~0.35‰)[48]、大洋中脊玄武岩和洋岛玄武岩的δ66Zn值范围(0.20‰~0.30‰)[49]相当, 也在目前报道的少数几个黄土和气溶胶的δ66Zn值(0.1‰~0.4‰)[48,50]数据范围内, 但小于台湾东部和南部海洋沉积物硅酸盐组分和台湾河流悬沙的δ66Zn值[51](图 5b)。这表明, 由于输入物源的差异,大陆边缘环境沉积物可能有变化范围较大的δ66Zn值。另外, 南海西部上升流区沉积物的δ66Zn值显著小于以生源硅为主和以碳酸盐为主的深海沉积物[48]。相比海洋悬浮颗粒物(0.15‰~0.43‰)[47]和浮游生物(0.08‰~0.57‰)[48,52], 南海西部上升流区沉积物以及大部分深海沉积物[48]的δ66Zn值略微偏小。
海洋沉积物的 Fe同位素组成受“输入海水的Fe端元同位素组成、水柱传输过程中的颗粒 Fe同位素组成变化、早期成岩过程对沉积物Fe同位素组成的影响”控制。输入边缘海的主要Fe端元有: 气溶胶、陆架孔隙水和河水, 相应的δ56Fe范围分别为0‰~0.2‰[43]、−2.5‰~−3.5‰[53]和−0.8‰~0.1‰[45]。边缘海水柱传输过程中的颗粒 Fe同位素组成变化尚无报道, 然而, 南海西部上升流区颗粒Fe同位素和痕量元素组成(据祁昌实未发表资料)表明, 深层水的颗粒 Fe同位素为铝硅酸盐 Fe同位素信号, 这意味着: 颗粒物水柱循环过程产生的Fe同位素信号并不能大量输出到海底沉积物。海洋沉积物早期成岩过程异化 Fe还原反应 Fe同位素分馏因子δ56FeFe(II)aq-Fe(III)reactive(约−1.3‰)[19,54,55]较大, 若有大量 Fe参与异化 Fe还原反应并发生迁移, 海洋沉积物Fe同位素组成将表现偏负(相对于成岩作用前)的趋势。以上研究表明, 南海西部上升流区沉积物的Fe同位素组成主要受“输入海水的 Fe端元同位素组成”和“早期成岩过程”控制。由于南海西部上升流区沉积物的δ56Fe值与黄土、气溶胶及火成岩一致, 与而静海环境沉积物有明显区别, 表明该上升流区缺乏来自邻近陆架孔隙水的轻同位素 Fe大量输入, 沉积物内部的异化 Fe还原反应强度较弱且(或)发生循环迁移的Fe比例不大, 也说明, 1~2 ka以来, 南海是一氧化性的海盆, 不存在周期性深部静水环境历史。
由于吸附/解吸附反应和有机物质降解控制着早期成岩过程中Zn的迁移循环[6], 所以除物源输入差异外, 最可能引起海洋沉积物Zn同位素变化的因素是: 吸附的Zn和有机质降解的Zn的同位素组成不同于海洋沉积物其余Zn组分、比例较大且发生循环迁移。生物通常优先利用偏轻的Zn同位素[52], 而吸附过程通常优先吸附较重的同位素[51–52]。除发生迁移的Zn比例可能较小原因外, 有机质降解和解吸附反应将分别释放偏轻和偏重的Zn同位素, 也可能导致成岩过程对海洋沉积物的 Zn同位素影响有限,从而使得目前观测到的包括南海西部上升流区沉积物在内的多数海洋沉积物样品的δ66Zn分布范围非常有限与黄土、气溶胶等陆源输出端元一致。
4 结 语
建立了适用于海洋沉积物的化学前处理及 Fe和Zn同位素测定方法, 该前处理方法可以实现海洋沉积物的快速消解、有机质去除和海盐脱离, 避免Fe和 Zn的意外丢失和由此导致的同位素分馏, 能够实现海洋沉积物Fe和Zn同位素精确测定。来自南海西部上升流海区两个沉积物柱样Y32和Y20的Fe和Zn同位素分析结果显示, 它们各自的δ56Fe值和δ66Zn值随深度无明显变化, 两个柱样之间也无明显差别, 与报道的黄土和气溶胶、火成岩以及大部分海洋沉积物一致。
厦门大学近海海洋环境科学国家重点实验室的林建荣、司靖宇和李骞帮助进行了相关样品采集;中国科学院地质与地球物理研究所岩石圈演化国家重点实验室的张艳斌博士和谢烈文高级工程师帮助进行了光谱纯NaCl的Cu-Fe-Zn含量测定; 两位匿名审稿人提出了宝贵的修改意见, 在此一并致以诚挚的谢意!
:
[1]Morel F M M, Milligan A J, Saito M A.Marine bioinorganic chemistry: The role of trace metals in the oceanic cycles of major nutrients[M]//Elderfield H.Treatise on Geochemistry(vol.6): The Oceans and Marine Geochemistry.Oxford:Elsevier-Pergamon, 2003: 113−143.
[2]Morel F M M, Price N M.The biogeochemical cycles of trace metals in the oceans[J].Science, 2003, 300(5621): 944−947.
[3]Martin J H.Glacial-interglacial CO2change: The iron hypothesis[J].Paleoceanography, 1990, 5(1): 1−13.
[4]Morel, F M M, Reinfelder J R, Roberts S B, Chamberlain C P,Lee J G, Yee D.Zinc and carbon co-limitation of marine phytoplankton[J].Nature, 1994, 369(6483): 740−742.
[5]Elrod V A, Berelson W M, Coale K H, Johnson K S.The flux of iron from continental shelf sediment: A missing source for global budgets[J].Geophys Res Lett, 31, L12307, doi:10.1029/2004GL020216.
[6]Santschi P H, Höehener P, Benoit G, Brink M B.Chemical processes at the sediment-water interface[J].Mar Chem, 1990,30: 269−315.
[7]Maréchal C N, Télouk P, Albarède F.Precise analysis of copper and zinc isotopic compositions by plasma-source mass spectrometry[J].Chem Geol, 1999, 156(1−4): 251−273.
[8]Belshaw N S, Zhu X K, Guo Y, O’Nions R K.High precision measurement of iron isotopes by plasma source mass spectrometry[J].Int J Mass Spectrom, 2000, 197(1−3):191−195.
[9]Schoenberg R, von Blanckenburg F.An assessment of the accuracy of stable Fe isotope ratio measurements on samples with organic and inorganic matrices by high-resolution multicollector ICP-MS[J].Int J Mass Spectrom, 2005,242(2/3): 257−272.
[10]Anbar A D, Rouxel O.Metal stable isotopes in Paleoceanography[J].Ann Rev Earth Planet Sci, 2006, 35:717−746.
[11]Dauphas N, Rouxel O.Mass spectrometry and natural variations of iron isotopes[J].Mass Spectrom Rev, 2006, 25:515− 550.
[12]Cloquet C, Carignan J, Lehmann M, Vanhaecke F.Variation in the isotopic composition of zinc in the natural environment and the use of zinc isotopes in biogeosciences: A review[J].Anal Bioanal Chem, 2008, 390(2): 451−463.
[13]李世珍, 朱祥坤, 唐索寒, 何学贤, 蔡俊军.多接收器等离子体质谱法Zn同位素比值的高精度测定[J].岩石矿物学杂志, 2008, 27(4): 273−278.Li Shi-zhen, Zhu Xiang-kun, Tang Suo-han, He Xue-xian, Cai Jun-jun.The application of MC-ICP-MS to high-precision measurement of Zn isotope ratios[J].Acta Petrol Mineral,2008, 27(4): 273−278 (in Chinese with English abstract).
[14]朱祥坤, 李志红, 赵新苗, 唐索寒, 何学贤, Belshaw N S.铁同位素的MC-ICPMS测定方法与地质标准物质的铁同位素组成[J].岩石矿物学杂志, 2008, 27(4): 263−272.Zhu Xiang-kun, Li Zhi-hong, Zhao Xin-miao, Tang Suo-han,He Xue-xian, Belshaw N S.High-precision measurements of Fe isotopes using MC-ICP-MS and Fe isotope compositions of geological reference materials[J].Acta Petrol Mineral, 2008,27(4): 263−272 (in Chinese with English abstract).
[15]Dauphas N, Pourmand A, Teng F.-Z.Routine isotopic analysis of iron by HR-MC-ICPMS: How precise and how accurate?[J].Chem Geol, 2009, 267(3/4): 175−184.
[16]Zhu X K, Guo Y, Williams R J P, O’Nions R K, Matthews A,Belshaw N S, Canters G W, de Waal E C, Weser U, Burgess B K, Salvato B.Mass fractionation processes of transition metal isotopes[J].Earth Planet Sci Lett, 2002, 200(1/2): 47−62.
[17]Welch S A, Beard B L, Johnson C M, Brateman P S.Kinetic and equilibrium Fe isotope fractionation between aqueous Fe(II) and Fe(III)[J].Geochim Cosmochim Acta, 2003, 67(22):4231−4250.
[18]Brantley S L, Liermann L J, Guynn R L, Anbar A D, Icopini G A, Baring J.Fe isotopic fractionation during mineral dissolution with and without bacteria[J].Geochim Cosmochim Acta, 2004, 68(15): 3189−3204.
[19]Icopini G A, Anbar A D, Ruebush S S, Tien M, Brantley S L.Iron isotope fractionation during microbial reduction of iron:The importance of adsorption[J].Geology, 2004, 32(3):205−208.
[20]Balci N, Bullen T D, Witte-Lien K, Shanks W C, Motelica M,Mandernack K W.Iron isotope fractionation during microbially stimulated Fe(II) oxidation and Fe(III)precipitation[J].Geochim Cosmochim Acta, 2006, 70(3):622−639.
[21]Maréchal C N, Sheppard S M F.Isotopic fractionation of Cu and Zn between chloride and nitrate solutions and malachite or smithsonite at 30℃ and 50℃[J].Geochim Cosmochim Acta, 2002, 66(suppl 1): A484.
[22]Rodushkin I, Stenberg A, Andrén H, Malinovsky D, Baxter D C.Isotopic fractionation during diffusion of transition metal ions in solution[J].Anal Chem, 2004, 76(7): 2148−2151.
[23]John S G, Geis R W, Saito M A, Boyle E A.Zinc isotope fractionation during high-affinity and low-affinity Zinc transport by the marine diatom Thalassiosira oceanica[J].Limnol Oceanogr, 2007, 52(6): 2710−2714.
[24]Balistrieri L S, Borrok D M, Wanty R B, Ridley W I.Fractionation of Cu and Zn isotopes during adsorption onto amorphous Fe(III) oxyhydroxide: Experimental mixing of acid rock drainage and ambient river water[J].Geochim Cosmochim Acta, 2008, 72(2): 311−328.
[25]Fehr M A, Andersson P S, Hålenius U, Mörth C-M.Iron isotope variations in Holocene sediments of the Gotland Deep,Baltic Sea[J].Geochim Cosmochim Acta, 2008, 72(3):807−826.
[26]Severmann S, Lyons T W, Anbar A, McManus J, Gordon G.Modern iron isotope perspective on the benthic iron shuttle and the redox evolution of ancient oceans[J].Geology, 2008,36(6): 487−490.
[27]Albarède F, Beard B L.Analytical methods for non-traditional isotopes[J].Rev Mineral Geochem, 2004, 55(1): 113−152.
[28]Murray J W.The interaction of metal ions at the manganese dioxide-solution interface[J].Geochim Cosmochim Acta,1975, 39(4): 505−519.
[29]Dawson K D, Donahoe R J.Zinc adsorption on Fe(II)/Fe(III)substates produced by microbial reduction of high-surfacearea goethite, medium-surface-area goethite and lepidocrocite[C]//AGU: AGU 2001 Fall Meeting, 2001:abstract #B32B−0114.
[30]司靖宇.金汞齐微电极的研制及其在测定海洋沉积物间隙水中DO、Mn2+和Fe2+的应用[D].厦门: 厦门大学, 2008.Si Jing-yu.Development of Hg-Au microelectrodes for measuring DO, Mn2+and Fe2+in pore water of marine sediments[D].Xiamen: Xiamen University, 2008 (in Chinese with English abstract).
[31]李建如.低纬海区碳储库与季风气候的轨道周期[D].上海:同济大学, 2007.Li Jian-ru.Carbon reservoir in low-latitude oceans and orbital cycles of monsoon climate[D].Shanghai: Tongji University,2007 (in Chinese with English abstract).
[32]唐索寒, 朱祥坤, 蔡俊军, 李世珍, 何学贤, 王进辉.用于多接收器等离子体质谱铜铁锌同位素测定的离子交换分离方法[J].岩矿测试, 2006, 25(1): 5−8.Tang Suo-han, Zhu Xiang-kun, Cai Jun-jun, Li Shi-zhen, He Xue-xian, Wang Jin-hui.Chromatographic separation of Cu,Fe and Zn using AGMP-1 anion exchange resin for isotope determination by MC-ICPMS[J].Rock Mineral Analysis,2006, 25(1): 5−8 (in Chinese with English abstract).
[33]唐索寒, 朱祥坤.AGMP-1阴离子交换树脂元素分离方法研究[J].高校地质学报, 2006, 12(3): 398−403.Tang Suo-han, Zhu Xiang-kun.Seperation of some elements using AGMP-1 anion exchange resin[J].Geol J China Univ,2006, 12(3): 398−403 (in Chinese with English abstract).
[34]Chapman J B, Mason T F D, Weiss D J, Coles B J, Wilkinson J J.Chemical separation and isotopic variations of Cu and Zn from five geological reference materials[J].Geost Geoanal Res, 2006, 30(1): 5−16.
[35]Mason T F D, Weiss D J, Horstwood M, Parrish R R, Russell S S, Mullane E, Coles B J.High-precision Cu-Zn isotope analysis by plasma source mass spectrometry: Part 1.Spectral interferences and their correction[J].J Anal At Spectrom,2004, 19(2): 209−217.
[36]Rosman K J R, Taylor P D P.Isotopic compositions of the elements 1997[J].Pure Appl Chem, 1998, 70(1): 217−236.
[37]唐索寒, 朱祥坤, 李津, 闫斌.地质样品铜、铁、锌同位素标准物质的研制[J].岩石矿物学杂志, 2008, 27(4): 279−284.Tang Suo-han, Zhu Xiang-kun, Li Jin, Yan Bin.Preparation of reference material for Cu, Fe and Zn isotope measurement of geological samples[J].Acta Petrol Mineral, 2008, 27(4):279−284 (in Chinese with English abstract).
[38]Zhao X, Zhang H, Zhu X, Tang S, Yan B.Iron isotope evidence for multistage melt-peridotite interactions in the lithospheric mantle of eastern China[J].Chem Geol, 2012,292−293: 127−139.
[39]Craddock P R, Dauphas N.Iron isotopic compositions of geological reference materials and chondrites[J].Geostandard Geoanal Res, 2010, 35(1): 101−123.
[40]Huang F, Zhang Z, Lundstrom C C, Zhi X.Iron and magnesium isotopic compositions of peridotite from Eastern China[J].Geochim Cosmochim Acta, 2011, 75(12): 3318−3334.
[41]Bigalke M, Weyer S, Kobza J, Wilcke W.Stable Cu and Zn isotope ratios as tracers of sources and transport of Cu and Zn in contaminated soil[J].Geochim Cosmochim Acta, 2010,74(23): 6801−6813.
[42]Severmann S, Johnson C M, Beard B L, McManus J.The effect of early diagenesis on the Fe isotope compositions of porewaters and authigenic minerals in continental margin sediments[J].Geochim Cosmochim Acta, 2006, 70(8):2006−2022.
[43]Beard B L, Johnson C M, Von Damm K L, Poulson R L.Iron isotope constraints on Fe cycling and mass balance in oxygenated Earth oceans[J].Geology, 2003, 31(7): 629−632.
[44]Rouxel O, Dobbek N, Ludden J, Fouquet Y.Iron isotope fractionation during oceanic crust alteration[J].Chem Geol,2003, 202(1): 155−182.
[45]Fantle M S, DePaolo D J.Irion isotopic fractionation during continental weathering[J].Earth Planet Sci Lett, 2004,228(3/4): 547−562.
[46]Zhu X K, O’Nions R K, Guo Y L, Reynolds B C.Secular variation of iron isotopes in north Atlantic deep water[J].Science, 2000, 287(5460): 2000−2002.
[47]Beard B L, Johnson C M, Skulan J L, Nealson K H, Cox L,Sun H.Application of Fe isotopes to tracing the geochemical and biological cycling of Fe[J].Chem Geol, 2003, 195(1-4):87−117.
[48]Maréchal C N, Nicolas E, Douchet C, Albarède F.Abundance of zinc isotopes as a marine biogeochemical tracer[J].Geochem Geophys Geosyst, 2000, 1(5), 1015, doi:10.1029/1999GC000029.
[49]Ben Othman D, Luck J M, Tchalikian A, Albarède F.Cu-Zn isotope systematics in terrestrial basalts (abstr)[C].Nice:Geophys Res Abstr (Europ Geophys Soc), 2003, 5: 9669.
[50]Luck J M, Ben Othman D, Albarède F, Telouk P.Pb, Zn and Cu isotopic variations and trace elements in rain//[C]Ármannsson H.Geochemistry of the Earth's Surface:Proceedings of the 5th International Symposium.Reykjavik:Taylor & Francis Group, 1999: 199−202.
[51]Bentahila Y, Ben Othman D, Luck J-M.Strontium, lead and zinc isotopes in marine cores as tracers of sedimentary provenance: A case study around Tarwan orogen[J].Chem Geol, 2008, 248(1/2): 62−82.
[52]John S G.The marine biogeochemistry of zinc isotopes[D].Woods Hole: M.I.T.and the WHOI, 2007.
[53]Severmann S, McManus J, Berelson W, Hammond D.The continental shelf benthic iron flux and its isotope composition[J].Geochim Cosmochim Acta, 2010, 74(14):3984−4004.
[54]Beard B L, Johnson C M, Cox L, Sun H, Nealson K H,Aguilar C.Iron isotope biosignatures[J].Science, 1999,285(5435): 1889−1892.
[55]Johnson C M, Beard B L, Roden E E, Newmann D K, Nealson K H.Isotopic constraints on biogeochemical cycling of Fe[J].Rev Mineral Geochem, 2004, 55(1): 359−408.
