饱和液态制冷剂氨的热物理性质的分子动力学模拟
2012-08-03赵胜喜祁影霞周生平
赵胜喜 祁影霞 周生平 张 华
(上海理工大学能源与动力工程学院 上海 200093)
氨是最早用于人工制冷的天然制冷剂之一,它的ODP和GWP均为零,而且氨泄漏后能够被雨水吸收返回土壤成为农田的肥料,是一种对环境无害的绿色制冷剂,广泛应用于国内外大中型冷库及其它低温制冷设备。氨的热物理性质的研究至关重要。传统的研究制冷剂热物性的方法主要有状态方程拟合和实验测定,但是这些方法既费时、费力又复杂。在此,提出运用分子动力学方法模拟制冷剂氨的热物理性质。
分子模拟是依据统计力学基本原理,将一定数量的分子输入计算机内进行分子微观结构的测定和宏观性质计算的模拟方法。分子动力学模拟[1](Molecular Dynamics Simulation )是分子模拟中应用于预测物质的热物理性质中最常用的一种方法。1971年,Rahman A等人[2]首次运用分子动力学模拟了液态水的性质。1989年,Vega C[3]运用分子动力学模拟了制冷剂R152a的热物理性质,Lusting R[4]模拟了液态乙烷的性质。1993年,Lisal M[5-7]等人模拟了制冷剂R143a、R152a、R142b的热力学性质,并优化了替代制冷剂R32和R23的势能模型函数。1997年,Seiji Higashi[8]等人运用Lennard-Jones势能模型模拟了制冷剂R32液态的热物理性质。2003年,清华大学的余大启[9]等人运用二中心LJ分子嵌入沿偶极矩(2CLJD)的分子模型,对制冷剂R134a的PVT性质进行了模拟。基于这些研究,这里在前人发展的site-site势能模型的基础上,模拟了制冷剂氨的饱和液态热物理性质。
1 分子动力学的基本原理
分子动力学方法是按体系内部的内禀动力学规律来计算并确定位形的转变。考虑含有n个分子的运动体系,系统中的能量包括分子的动能与总势能的和。其总势能为分子中各原子位置的势能函数之和,一般用 表示。根据经典力学,系统中任一原子所受之力为势能的梯度:

由牛顿运动定律可得原子的加速度为:

将牛顿运动定律方程式对时间积分,可预测原子经过时间t后的速度与位置:
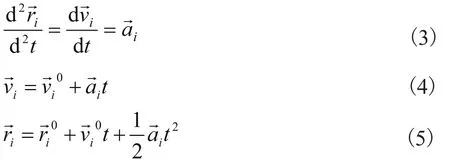
式中,U代表原子所受的势能。 和mi分别代表原子所受之力和质量, 和 分别代表原子的速度,加速度和位置。上标“0”代表各物理量的初始值。
由于加速度在时间上是连续函数,在计算中,我们通常把时间表达成离散的形式:

式中,tn是原子到第n步的时间;n是步数;h是时间步长,表示一段非常短的时间间隔(一般取0.8fs)。假定在时间步长h范围内粒子受到的力是不变的,给定粒子的初始位置和初始速度,则可以对该方程进行数值求解。计算过程如下图1所示:

图1 数值求解示意图Fig.1 Schematic diagram of numerical solution
通过以上的反复循环计算,可得到各时间下系统中各原子运动的位置、速度及加速度等数据。然后再利用统计计算方法得到系统的静态和动态特性,从而得到系统的宏观性质,比如压力,密度,内能,焓等热物理性质。
2 势能模型
氨的势能模型采用site-site势能模型[10],分子间的相互作用力包括Lennard-Jones(6-12)项和库仑作用项,其形式如下:

其中,r是不同分子中的原子间的距离,σ和ε分别是分子中不同原子间的L-J势能的能量和尺度参数。q是分子中各原子所带的部分电荷,ε0是自由介电常数,e是电荷的单位。下标a、b分别代表不同分子中的原子对。
对于氨分子中的不同原子的σ、ε和q可以运用量子力学的从头计算方法获得,模拟采用Bernhard Eckl[11]等人的优化结果,如表1所示。

表1 氨分子的势能参数Tab.1 Potential function parameters of ammonia
3 模拟方法
分子动力学计算之前,必须先估计计算的可行性,一般来说系统的原子个数越多,模拟的结果越精确,但是以目前的计算机水平,分子动力学计算的容量约为数千个原子的系统。此次模拟选用500个氨分子进行面心立方晶格建模,首先采用等温等容(NVT)系综使体系达到平衡,然后在等温等压 (NPT)系综下得到模拟结果。对建立的面心立方晶格模型采用周期性边界条件,利用弛豫作用随机得到分子的初始位置。针对各分子间的Lennard-Jones项采用硬球截断法,截断半径为立方晶体边长的一半,长程静电力则采用Ewald加和法计算。运动方程采用基于预测-校正积分方法的Gear算法,温度采用速度标定法控制。时间步长采用0.8fs, 模拟步程约为50000步,其中前30000步用于体系达到平衡,后20000步用于得到氨的热物理性质。
4 模拟结果
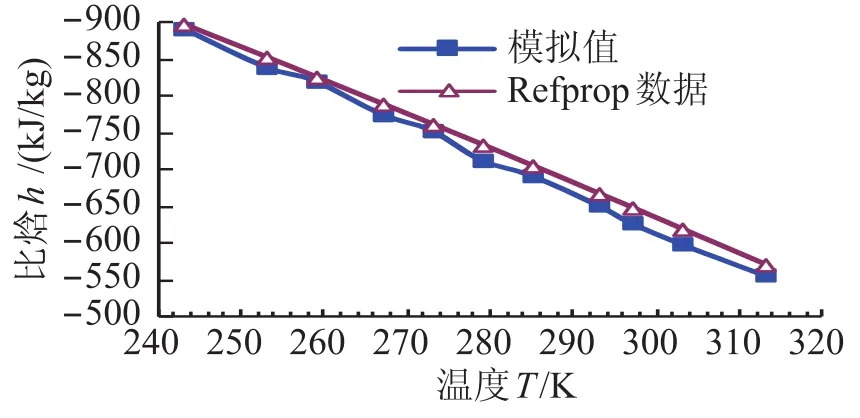
图2 制冷剂氨的饱和液态T-h图线Fig.2 Saturated liquid T-h of refrigerant ammonia
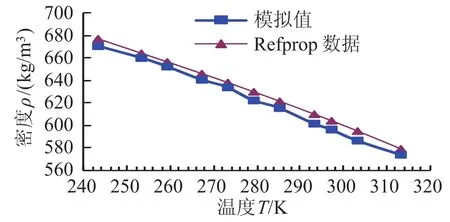
图3 制冷剂氨的饱和液态T-ρ图线Fig.3 Saturated liquid T-ρ of refrigerant ammonia
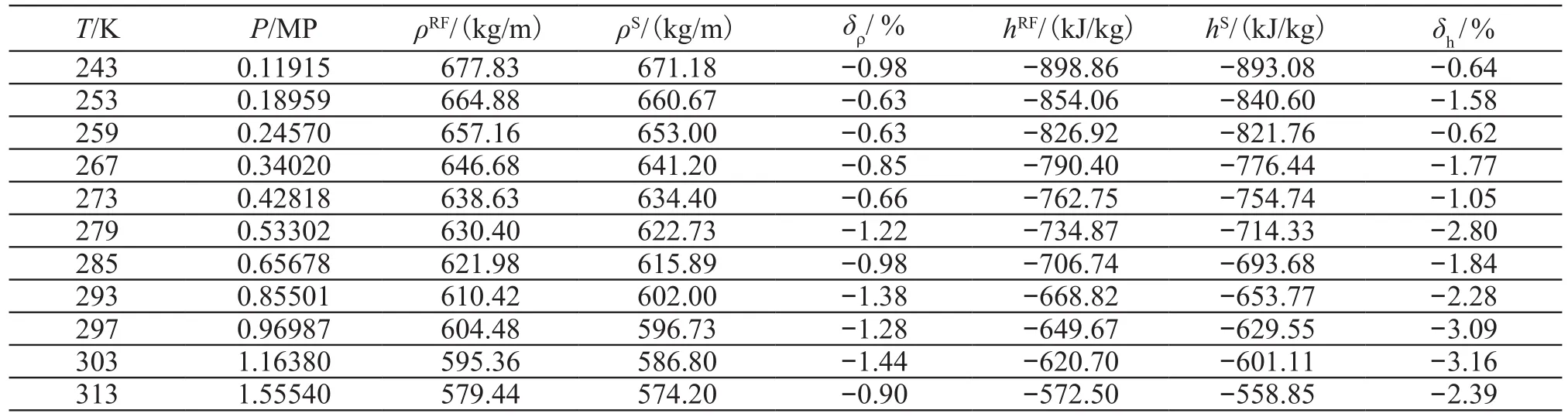
表2 制冷剂氨的饱和液态热物理性质Tab.2 Saturated liquid thermodynamic properties of refrigerant ammonia
运用分子动力学方法模拟了制冷剂氨的饱和液态热物性,并将模拟结果与美国国家标准研究所(NIST)的Refprop8.0数据库提供的制冷剂氨的饱和液态热物性质进行了比较,其中比焓是以0℃的饱和液态氨的比焓为基准得到(h=-762.75kJ/kg)。主要结果如表2,图2和图3所示。
从表2、图2及图3可知,模拟结果同美国国家标准研究所(NIST)的数据库提供的氨的饱和液态热物理性质有很好的一致性。氨饱和液态密度的最大相对偏差在1.5%以内,比焓的最大相对偏差在3.2%以内,低压区的模拟结果比高压区的模拟效果好。
5 结论
采用site-site势能模型对制冷剂氨的饱和液态密度和比焓进行了分子动力学模拟,模拟结果与NIST数据库的最大相对偏差分别在1.5%(密度)以内和3.2%(比焓)以内。表明采用合理的势能模型和参数,运用分子动力学方法来预测单一组分工质的热物理性质是可行的。
本文受上海市重点学科建设项目(S30503),上海市教育委员会科研创新项目(11YZ119)及上海市研究生创新基金项目(JWCXSL1102)资助。(The project was supported by Shanghai Leading Academic Discipline Project (No.S30503),Shanghai Education Commission Scienti fi c Research Innovation Projects (No.11YZ119) and The Innovation Fund Project For Graduate Student of Shanghai(No.JWCXSL1102).)
[1]陈正隆,徐为人,汤立达.分子模拟的理论与实践[M].北京:化学工业出版社,2007.
[2]Rahman A, Stillinger F H. Molecular dynamics study of liquid water [J]. Chem Phys, 1971, 55: 3336-3359.
[3]Vega C, Saager B, Fischer J. Molecular dynamics studies for the new refrigerant R152a with simple model potentials[J]. Molecular Physics, 1989, 68(5): 1079-1093.
[4]Lusting R, Toro-Labbe A, Steele W A. Molecular dynamics study of the thermodynamics of liquid ethane [J].Fluid Phase Equilibria, 1989, 48:1-7.
[5]Lisal M, Budinsky R, Vacek V, et al.Vapor-liquid equilibria of alternative refrigerants by molecular dynamics simulation [J].Int Thermophysics,1993,20(1):163-169.
[6]Lisal M, V Vacek. Effective potentials for liquid simulation of the alternative refrigerants HFC-32:CH2F2and HFC-23: CHF3[J]. Fluid Phase Equilibria, 1996, 118:61-76.
[7]Lisal M, Vacek V. Molecular dynamics simulations of fluorinated ethane [J]. Molecular Physics, 1996,87(1):167-187.
[8]Seiji Higashi,Akira Takada.Molecular dynamics study of liquid CH2F2(HFC-32)[J].Molecular Physics,1997,92:641-650.
[9]余大启, 李卓毅, 曹炳阳, 等. R134a PVT性质的分子动力学模拟[J].工程热物理学报,2003,24(6):24-26.(Yu Daqi, Li Zhuoyi,Cao Bingyang, et al.Molecular dynamics studies about the PVT properties of R134a[J].Journal of Engineering Thermophysics,2003,24(6):24-26.)
[10]T Kristof, J Vorholz, J Liszi, et al. A simple effective pair potential for the molecular simulation of the thermodynamic properties of ammonia [J]. Molecular Physics, 1997, 97: 1129-1137.
[11]Bernhard Eckl, Martin Horsch, Jadran Vrabec, et al.Molecular modeling and simulation of thermophysical properties: application to pure substances and mixtures[G]// High Performance Computing in Science and Engineering'08, 2009,3:119-133.
