左冠状动脉异常起源于肺动脉的心电图特点及术后转归
2012-05-23冯雪李晓峰袁越张励兵宋振江
冯雪,李晓峰,袁越,张励兵,宋振江
左冠状动脉异常起源于肺动脉(ALCAPA),也称为Bland-White-Garland综合征,是一种极罕见的先天性心血管畸形,发病率为1/300,000个活产婴儿,约占先天性心脏病的0.25% ~0.5%[1]。由于发病率低,临床表现缺乏特异性,极易导致误诊或漏诊,错过手术治疗。据文献报道,ALCAPA的患儿未及时诊治,预后极差,1岁内死亡率高达90%[2]。近年来,随着临床医生及影像学诊断水平的提高,ALCAPA的确诊率得到一定的提高。本文对12例确诊ALCAPA术前心电图和外科治疗术后随访12个月的心电图进行回顾性分析,旨在探讨ALCAPA的心电图诊断规律和术后异常Q波变化,以及提高对该病的认识。
1 资料与方法
对象:从2006-01至2011-12我院心脏外科总共实施先天性心脏病手术2015例,其中12例经超声心动图、心血管造影及增强计算机断层摄影术(CT)扫描诊断为ALCAPA,并通过手术证实。根据手术时年龄分为婴儿组(≤1岁,n=6)和儿童组(>1岁且≤5岁,n=6)。
方法:(1)临床资料分析:①术前临床症状;②既往有无心功能不全史。(2)心电图分析:①术前(最近一次)和术后随访(1、6、12个月)应用日本光电赛维亚牌心电图检测仪对本组病例行标准12导联心电图检查。②主要分析以下心电图特征:Ⅰ导联异常Q波和T波倒置;aVL导联异常Q波和T波倒置;V4~6导联异常Q波和ST-T改变;左心室肥厚。③心电图异常Q波定义为Q波深度超过R波的1/4(Q/R>0.25)、左心前导联Q/R>0.15且时程≥0.03 s;ST段压低是指ST段下移≥0.05 mV;左心室肥厚标准按现行规定[3]。④术后心电图随访指标:针对异常Q波(Ⅰ、aVL、V4~6导联)变化分为:缓解:3个导联异常Q波消失;部分缓解:1或2个导联异常Q波消失;无变化:3个导联异常Q波仍存在。(3)超声心动图分析:①本研究病例行二维彩色多普勒超声心动图检查,使用仪器型号为Philips sonos 5500或iE33;②左心室收缩功能评估指标:按照射血分数(EF)分为正常(EF≥60%)、减低(50%≤EF<60%)、差(EF<50%);③术前(最后一次)及术后随访(1、6、12个月)超声心动图,了解左心室收缩功能情况。(4)手术治疗:12例均在全麻插管、体外循环心内直视下行外科手术治疗。8例采取左冠状动脉(LCA)直接移植术,4例采用肺动脉内隧道术(Takeuchi术),以达到异常起源于肺动脉的LCA开口与主动脉根部的连接,恢复心肌顺行的双冠状动脉系统血供。
统计学方法:采用SPSS 11.0统计软件进行分析。对婴儿组和儿童组术前临床表现、心电图特征的比较采用确切概率检验(Fisher’s exact test);对两组术后心电图异常Q波变化的随访比较采用两组等级资料的秩和检验。P<0.05为差异有统计学意义。
2 结果
两组ALCAPA患儿的临床表现:①婴儿组6例,男2例,女4例,年龄1个月 ~8个月(平均4.3个月),体重3.0 kg~11.8 kg(平均6.2 kg);儿童组6例,男2例,女4例,年龄1.2岁 ~4.5岁(平均4.3岁),体重7 kg~22 kg(平均12.5 kg)。②婴儿组6例患儿就诊时均表现为不同程度的气促、喘憋症状,均有喂养困难和活动耐受力减低等心功能不全史,儿童组中仅1例有上述症状。
术前心电图和超声心动图:(1)心电图:①在Ⅰ、aVL、V4~6导联中出现异常Q波:婴儿组分别为5例、5例和6例,儿童组分别3例、4例和1例,婴儿组均高于儿童组,只有V4~6导联两组比较差异有统计学意义(P<0.05)。②Ⅰ、aVL 导联 T 波倒置及 V4~6导联 ST-T改变:婴儿组6例均出现,儿童组分别为4例、5例和3例;左心室肥厚:婴儿组6例,儿童组4例,上述导联心电图改变婴儿组均高于儿童组,但差异无统计学意义(P>0.05)。(2)超声心动图左心室收缩功能(EF值):婴儿组EF值[(44.3±8.9)%]明显低于儿童组EF值[(57.4±6.5)%],差异有统计学意义(P<0.05)。
术后心电图和超声心动图:对术后9例存活的ALCAPA(婴儿组3例,儿童组6例)进行1、6、12个月随访(1)心电图:①术后1个月,Ⅰ、aVL、V4~6导联异常Q波婴儿组3例,与术前无变化;儿童组1例在三个导联仍可见异常Q波,5例呈现部分缓解,两组差异有统计学意义(P<0.05);②术后6个月,婴儿组2例在三个导联可见异常Q波,1例部分缓解;儿童组4例部分缓解,2例三个导联异常Q波消失、呈缓解情况,两组差异有统计学意义(P<0.05);③术后12个月,婴儿组1例在三个导联异常Q波仍然存在,2例部分缓解;儿童组1例在aVL导联可见异常Q波,5例三个导联异常Q波全部消失、呈缓解情况,两组差异有统计学意义(P<0.05),图1。(2)超声心动图:①术后1个月,婴儿组 EF值[(46.2±5.1)%],儿童组 EF值[(57.9±4.8)%],与术前比较差异均无统计学意义(P>0.05);②术后6个月和12个月,婴儿组EF值分别为(53.1±4.8)%和(59.4±3.3)%,均较术前明显改善,差异均有统计学意义(P=0.043和P=0.037);儿童组EF值分别为(59.2±5.3)%和(62.3±2.7)%,较术前进一步改善,逐步接近或达到正常,但差异无统计学意义(P均>0.05)。
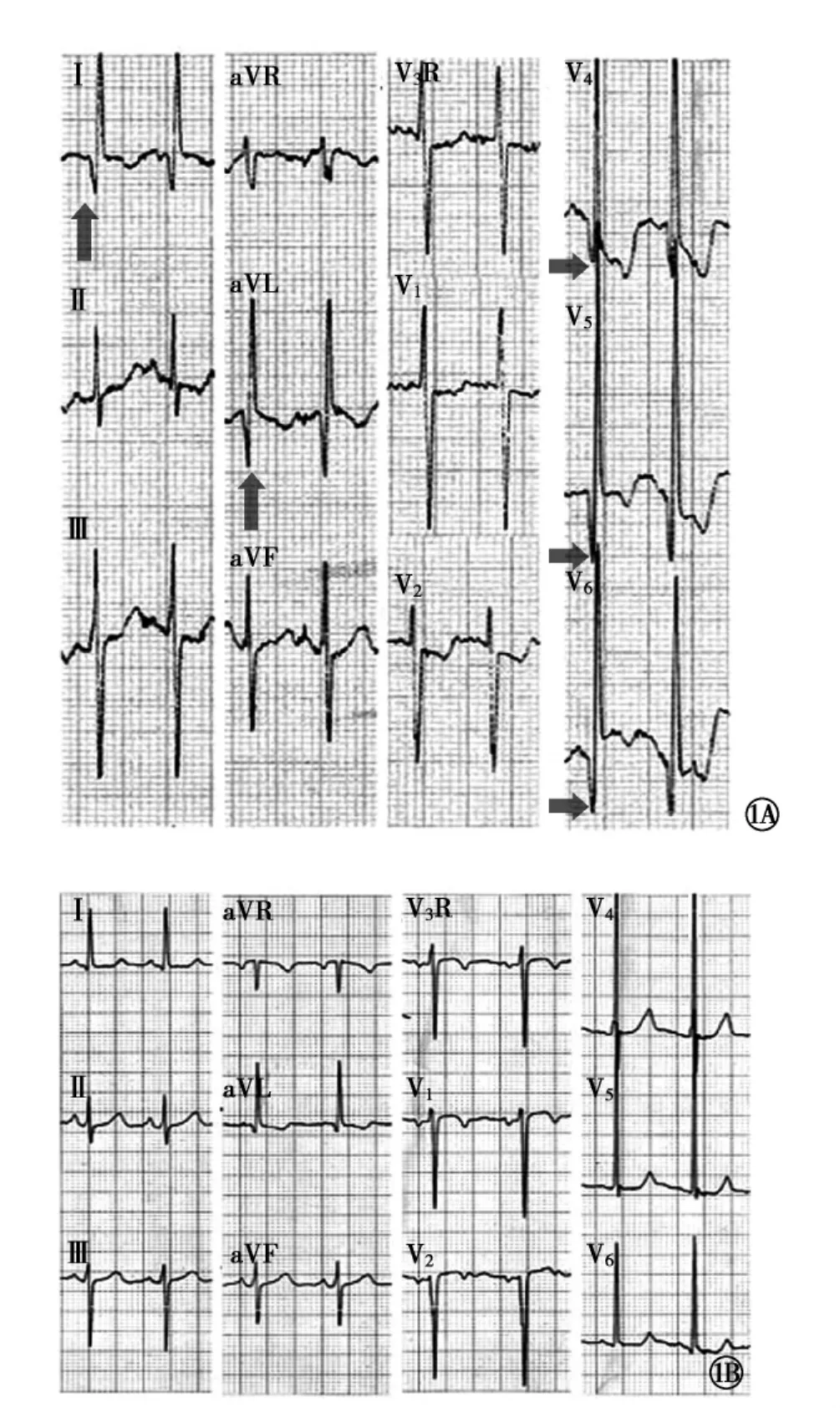
图1 左冠状动脉异常起源于肺动脉病例术前和术后心电图变化1A术前Ⅰ、aVL和V4~6导联显示异常Q波(箭头);1B术后12个月Ⅰ、aVL和V4~6导联异常Q波完全消失
手术所见及治疗结果 ①12例ALCAPA经手术后得到证实,其中合并动脉导管未闭4例,室壁瘤1例,右心室流出道狭窄1例,其余均为单发畸形。12例患儿均有侧支循环形成,且都表现为右冠状动脉(RCA)迂曲扩张;儿童组均有丰富的侧支血管及血管网络形成,婴儿组只有少量侧支血管存在。12例患儿术中未见明显心肌梗塞灶,婴儿组4例和儿童组1例可见左心室壁局部纤维化、心肌成灰白色。②12例手术患儿死亡3例(术后2周内),存活9例;死亡3例均来自婴儿组,死亡原因为围手术期低心排,术前左心室收缩功能差(EF值均低于40%),术前心电图改变无明显特异性。
3 讨论
冠状动脉异常起源于肺动脉是一种罕见的先天性心脏病,发病率约占先天性心脏病的0.4%[4]。正常情况下,在胚胎发育第9周时由血管母细胞芽形成冠状动脉系统远端,并穿过心内膜形成大的冠状动脉分支。近端冠状动脉在动脉干附近形成一个环,与原始主动脉窦处的冠状动脉芽连接一起作为动脉干部分形成大动脉。若近端部分在形成过程中发生移位,即可导致冠状动脉异常起源于肺动脉。ALCAPA临床最多见,约占73.3% ~77.4%[5]。随出生后肺循环系统压力下降、左冠状动脉血流灌注减低,如果缺乏良好的右冠状动脉与左冠状动脉之间的侧支循环,早期可形成严重的左心室心肌缺血、梗死,并发展心力衰竭,临床经常表现为气促、喘憋、喂养困难等,多数患儿1岁内死亡[6];如果自右冠状动脉到左冠状动脉的侧支循环建立良好,足以维持左冠状动脉的灌注压和血流量,一般无临床症状,少数可以存活至青少年甚至成年期[7,8]。本文结果显示,婴儿组6例均存在心功能不全的表现,明显高于儿童组的1例;经过心脏手术证实婴儿组侧支循环较儿童组明显不足,与文献报道相吻合[9,10]。
ALCAPA的心电图常有特征性表现,主要为前侧壁心肌梗死的改变,即Ⅰ、aVL、V4~6导联的异常Q波、T波倒置和ST-T改变,而Ⅰ、aVL导联深而宽的Q波伴T波倒置经常是本病的诊断要点[7],同时对鉴别扩张型心肌病和心内膜弹力纤维增生症有重要意义[11,12]。本文数据显示,两组在Ⅰ、aVL 导联均可见异常Q波伴T波倒置,符合ALCAPA心电图特点;但是在V4~6导联,婴儿组有明显异常Q波,与儿童组有显著性差异,以此可以作为婴儿患者重要的诊断依据,同时对婴儿与儿童两组患者间相互鉴别具有诊断价值。
ALCAPA是小儿最常见心肌缺血或梗死性疾病之一,自然预后和内科治疗效果差,婴儿期死亡率非常高。通过尽早手术治疗,恢复心肌顺行双条冠状动脉血流灌注,缓解左心室心肌缺血性损伤,可以使左心室收缩功能逐渐恢复至正常,提高生存率。有文献报道[13],对ALCAPA的患者实施手术治疗,恢复正常两条冠状动脉血流循环之后,左心室收缩功能不全和二尖瓣反流可以逐渐改善,同时病理性Q波也随之慢慢部分缓解、最终完全消失。从本文对9例ALCAPA术后存活的病例进行为期12个月的心电图和超声心动图随访结果,可以得出如下结论:(1)心电图方面:①术后1、6、12个月,异常 Q波呈现逐渐部分缓解,婴儿组分别为0例、1例和2例;儿童组分别为5例、4例和1例;②术后1、6、12个月,异常Q波可以逐渐消失,婴儿组3例在1~3个导联仍可见,儿童组异常Q波消失数分别为0例、2例和5例;③儿童组异常Q波得到部分缓解和消失的时间较婴儿组明显提前、恢复更快、效果更佳。(2)左心室收缩功能变化:①术后短期内(1个月),两组EF值改善不明显;②术后6、12个月,婴儿组的EF值明显提高、甚至接近正常;儿童组也有一定程度提高,逐渐达到正常;③术后婴儿组左心室收缩功能改善程度较儿童组更明显。由于婴儿患者左右心室之间有效侧支循环少,所以术前心电图的心肌缺血和梗死性改变显著、异常Q波明显、左心室收缩功能差;在儿童患者,由于存在较丰富的侧支循环,所以术前上述心电图改变和左心功能减低表现得不严重。另外术中可见心肌有缺血改变,没有发现心肌梗塞或梗死的病灶,有别于成人的心肌梗死。从本文随访结果显示:①ALCAPA患儿心肌缺血性改变具有可逆性,对确诊的ALCAPA患儿(尤其是小婴儿)尽早成功实施冠状动脉移植手术,有利于恢复左心室心肌正常冠状动脉血流供应,促使心肌缺血性损伤逐渐缓解,达到异常Q波逐渐消失和左心室收缩功能改善;②婴儿组异常Q波消失速度较儿童组慢,但是左心室收缩功能改善程度较儿童组明显;③伴随术后异常Q波逐渐消失,两组患儿左心功能逐渐恢复,但是异常Q波消失是一个逐渐过程、需要相对较长的时间,临床左心功能改善较快。
综上所述,对于小儿临床表现心功能不全、心脏增大的病例,心电图Ⅰ、aVL导联出现异常Q波和T波倒置对本病的诊断有重要提示作用;在V4~6导联(左胸前导联),心电图有明显异常Q波存在,对于诊断婴儿型ALCAPA有较高的灵敏性和一定的特异性,可以作为与儿童型鉴别的重要依据;对小婴儿早期实施外科手术治疗,有利于恢复左心室正常冠状动脉血流、促使异常Q波逐渐消失和左心功能不全纠正,降低死亡率。心电图医师应该加强对本病的认识,重视心电图改变,同时结合心脏超声、心脏CT及心血管造影结果,对提高ALCAPA检出率、减少误诊具有极其重要的意义。
[1]Dodge-Khatami A,Mavroudis C,Backer CL.Anomalous origin of the left coronary artery from the pulmonary artery:collective review of surgical theapy.Ann Thorac Surg,2002,74:946-955.
[2]Wesselhoeft H,Fawcett JS,Johnson AL.Anomalous origin of the left coronary artery from the pulmonary trunk.Circulation,1968,38:403-425.
[3]梁翊常主编.实用小儿心电图学.第二版.北京:人民卫生出版社,1998.61-62.
[4]Friedman WF,Silverman N.Congenital heart disease in infancy and childhood[M]in:Braunwald E,Zipes DP,Libby P.Heart Disease:a textbook of cardiovascular medicine.6th.Philadelphia:WB.Saunders,2001.1505-1591.
[5]Garg N,Tewari S,Kapoor A,et al.Primary congenital anomalies of the coronary arteries:a coronary arteriographic study.Int J Cardiol,2000,74(1):39-46.
[6]许光,杨敏富,吕小东,等.应用18F-脱氧葡萄糖心肌显像对儿童左冠状动脉起源于肺动脉存活心肌的评价.中国循环杂志,2010,25(5):371-375.
[7]张萱,李鲁光,张桂珍,等.左冠状动脉起源于肺动脉一例.中国循环杂志,1999,14(2):74-75.
[8]汪周平,张丽,于明华,等.儿童冠状动脉起源异常的临床分析.广东医学,2010,31(13):1731-1733.
[9]Zheng JY,Han L,Ding WH,et al.Clinical features and long-term prognosis of patients with anomalous origin of the left coronary artery from pulmonary artery.Chin Med J,2010,123(20):2888-2894.
[10]Samuel R,Pedro JC,Juan CC,et al.Outcomes of coronary reimplantation for correction of anomalous originof left coronary artery from pulmonary artery.Rev Esp Cardiol,2011,64(8):681-687.
[11]郭保静,韩玲,金梅,等.心电图对婴儿型左冠状动脉起源于肺动脉的诊断价值.中华儿科杂志,2004,42:863-864.
[12]张辉,罗毅,尤斌,等.婴儿型左冠状动脉起源肺动脉的诊断治疗及外科治疗[J].中华胸心血管外科杂志,2005,21(6):372-373.
[13]Chiu HH,Wang JK,Chen CA,et al.Resolution of pathologic Q wave,left ventricular dysfunction and mitral regurgitation after dual coronary repair of the anomalous origin of the left coronary artery from the pulmonary artery.Eur J Pediatr,2008,167:1277-1282.
