标准化实时荧光定量PCR技术流程
2012-05-06曹龙凯郭春华
曹龙凯,郭春华
(西南民族大学生命科学与与技术学院,动物遗传育种国家民委-教育部重点试验室,成都 610041)
随着分子生物学的发展,实时荧光定量PCR(RT-qPCR)技术已经成为检测不同样品间基因表达水平定量差异的权威性方法,但是也出现了许多缺陷,包括试验设计不合理、对照和重复缺乏、分析方法和报告形式多样、RNA样品质量、引物和探针低劣选择等。实时荧光定量PCR试验出版物所需最小信息量(MIQE)是一套国际新推出的指导方针,对RT-qPCR的试验和发表文章所必需的信息提出了最低限度的标准[1]。MIQE与蛋白质组学试验、基因组序列、RNA干扰、代谢研究和NDA微阵列分析的国际标准一样,作为RT-qPCR的国际标准——MIEQ也被收入MIBBI[2-8]。MIQE对RT-qPCR的过程做了详尽的阐述,从而使科学工作者能获得一致的、高质量的RT-qPCR试验数据。按照这一标准,可以规范试验操作,提高研究者对试验结果的可信度,增强试验的透明度,促进试验室之间的一致性,进一步规范qPCR的仪器和试剂的研发生产[9-12]。
MIQE所提供的检测列表包含85个参数,满足了所有杂志的评审标准,确保了试验结果的质量,应对mRNA转录的高动态性和样品操作及下游处理步骤的多变性,使通过该标准评估的试验结果具有一致性、可靠性和可比性[1]。另外,还规范了RT-qPCR的专用术语,以统一实时定量PCR的试验数据[13]。
根据MIQE指南,一个典型的RT-qPCR试验流程包括试验设计、RNA提取、引物和探针设计、RT-PCR、数据分析、对照组、重复数。从试验条件到样品操作,都要进行精心的安排。本文介绍了一套标准化的RT-qPCR的试验方法,为RT-qPCR试验提供参考。
1 RT-qPCR试验设计
由于mRNA的超敏感,反应需要在严格的调控下进行,适当的试验设计是研究基因表达成功的关键,花一定的时间在试验设计上是很必要的。试验设计内容包括样品抽提方法、保存方法、解冻和均质化过程、对照组设置、靶基因和内参基因选择,生物学和技术重复次数等都要进行严格的考虑,从而可以减少试验结果的可变性,保证其顺利进行。
2 RNA提取
2.1 RNA提取的原理和方法
组织总RNA提取的实质就是将细胞裂解,释放出RNA,并通过RNA的纯化去除蛋白质、DNA等杂质,最终获得高纯度RNA的过程。
细胞裂解法有苯酚法和β-巯基乙醇法。苯酚法是一种传统的RNA提取方法,适用于大多数动植物材料,但对次生代谢产物较多的植物材料提取RNA效果较差;β-巯基乙醇法适用于各种不同动物材料和次生代谢物少的植物材料;对于次生代谢物多的材料,一些生物公司亦推出了RNA提取试剂,不同的提取方法适用于不同的样本材料,试验者可根据自己的需要进行选择。
RNA的纯化及沉淀方法包括有机溶剂抽提法和硅基质吸附法。有机溶剂抽提法通常用氯仿抽提RNA,去除蔗糖、蛋白质等杂质;硅基质吸附法是利用硅基质对核酸特异的吸附能力,该法因操作方便、快捷、不使用有毒的溶剂(苯酚、氯仿)等优点,成为了核酸纯化的首选。
现在的RNA提取试剂盒综合了上述细胞裂解和RNA纯化的一系列步骤,提取方便,易于操作,是一种理想的试剂盒。但若要提取一些特殊的样品,可选用专用的试剂盒,例如动物组织提取试剂盒,该类试剂盒比综合提取试剂盒效果更好。另外,在使用前应对试剂盒的质量进行检测,以达到预期的效果。
2.2 样品的预处理
样品的预处理方法要根据样品的不同来选择不同的方法,植物材料需要液氮研磨,动物材料需要匀浆、液氮研磨,细菌需要用溶酶菌破壁,酵母需要液氮研磨、玻璃珠处理或lyticase酶法破壁。如果样品在采集后无法立即处理,需要放置一段时间,应先将材料在液氮中速冻后保存于-80℃冰箱或直接保存在液氮中。
3 RNA质量的控制
RT-qPCR试验流程中,高纯度和高完整性的RNA是试验的关键。不纯的RNA样品会抑制PCR反应,产生偏离的数据;不完整的RNA会产生不正确和可变的定量结果[14]。RNA用于不同的后续试验,对其质量要求不尽相同,cDNA文库构建要求RNA完整且无酶等抑制物残留;Northern blot试验对RNA完整性要求较高,对酶反应抑制物要求较低;RT-PCR试验对RNA完整性要求不太高,但对酶反应抑制物残留要求严格。
3.1 RNA纯度的检测
采用分光光度法检测RNA的纯度,以OD260/230值作为参考值。OD260/280值为1.9~2.1,可以认为RNA纯度较好;OD260/280值<1.8,则表明有蛋白质杂质;OD260/280值>2.1,说明RNA已经降解;OD260/230值>2.0,则表明裂解液中有β-巯基乙醇和异硫氰酸胍残余。另外,采用TE溶解或洗脱RNA会使OD260/280值增加。总RNA浓度的计算公式见式(1):
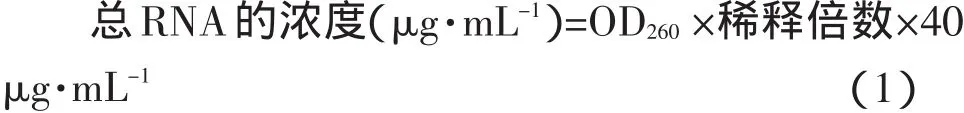
3.2 RNA完整性鉴定
根据后续试验的不同,对鉴定的要求也有所差异。一般可分为变性电泳和常规电泳,由于变性电泳操作步骤比较复杂,所以在不太严格的试验中,RNA的电泳可以用常规的1%琼脂糖凝胶电泳。总RNA在普通琼脂糖凝胶电泳上出现的条带与变性凝胶上的一致,可以观察到一大一小(28S和18S)两条带,其大条带的亮度是小条带亮度的200%,则提示RNA完整,这种方法成本低,但判断上具有一定主观性,并且需要总RNA 200 ng,如果样品稀少,则相对浪费。使用光密度扫描仪对rRNA条带的亮度进行扫描可以改善上述方法,虽然28S/18S的值在不同组织中有所变动,不过比值为1~2,则说明RNA样品的完整性好。
然而对RNA完整性要求较高的后续试验,如Northern blot和cDNA文库的构建,则需要通过微流体电泳系统,对RNA的完整性做出客观的、准确的评判[15]。该系统是目前对RNA的浓度质量(从pg到ng级)和完整性的检测最完善的系统,可以生成一块虚拟胶,基于电泳图的多个标准,也可以提供RNA样品的客观判断以及降解样品的筛选[16]。
综上所述,通过确保RNA的纯度和质量的一致性可以降低生物学重复的差异[15]。建议在RNA样品符合标准时应立即进行RT-PCR,以便在检测后保持其完整性。
4 引物和探针设计
引物设计是PCR步骤中十分关键的一步,引物的好坏决定着试验的成功与否。对于RT-qPCR,引物设计和目标序列选择的要求就更严格了。依照MIQE指南,荧光定量PCR的引物必须是以两个外显子设计引物,避免基因组DNA的扩增,且在发表文章中必须提供每个目的基因和对照基因的da⁃tabase检索编号,引物和探针的外显子位置。
设计引物应注意先选择好探针,然后再设计引物,使之尽可能的靠近探针;所选序列应是高特异的,尽量选择最小二级结构的扩增片段,设计时应先检验,若无法避免就要相应提高退火温度;扩增片段的长度应<400 bp,理想的是100~150 bp,太短也有其弊端,如影响片段回收;GC含量应在50%~65%,GC含量太高,影响引物的退火温度,其容易产生非特异性扩增,从而影响扩展增效率及产生非特异信号;尽量避免连续的G或C出现;引物和探针配检,以避免二级结构和发夹结构的产生;合成好的探针,在用之前要先离心,用双蒸水(灭菌)稀释成25 pmol·μL-1的浓度,然后于-20℃避光保存。
设计引物需要的软件有Primer 6.0(设计夸外显子引物)、Primer 5.0(检测上下游引物的错配)、Ol⁃ige 6.0(检测上、下引物的二级结构和发夹结构出现能值的高低)。另外,设计好的引物还要通过NCBI中的Primer-Blast来进行比对,以确保目的基因的特异性。MFOLD程序可用来分析扩增子是否存在阻碍有效扩增的二级机构[17]。
5 反转录、扩增产物的验证和RT-qPCR扩增
反转录和RT-qPCR扩增中,MIEQ规定聚合酶的浓度和性质、cDNA的量、镁离子浓度、buffer的化学成分、反应体积以及RT-qPCR仪的循环条件都必须注明。
5.1 反转录
由于RNA酶在环境中广泛存在,RNA提取完成后,应尽快进行反转录以避免RNA样品在反转录之前反复冻融而导致RNA样品的降解。在确保RNA质量后,即可进行反转录。在引物的选择方面,根据RT引物数据库且视试验的具体情况而定,选择随机引物、Oligo dT及基因特异性引物,对于短的不具有发夹机构的真核细胞mRNA,3种引物都可以[18-19]。
RT-PCR分为一步法和两步法,可以根据不同目的选用不同的方法。若要达到高灵敏度的效果,建议用一步法;如果RT-PCR使用不同的引物,同时要得到高特异、高保真、长的反转录结果,就采用两步法。此外,MIQE中对反转录过程要求设置无转录(NRT)对照。
5.2 扩增产物的验证
RT-qPCR之前,要对引物进行验证,验证扩增产物是否是目的片段。具体做法是:做普通PCR,然后胶回收产物,连接到质粒上;对质粒进行测序,确定是目的片段。
5.3 RT-qPCR扩增与验证
RT-qPCR扩增流程中,PCR的效率、线性动态范围、退火温度、融解曲线、扩增子的凝胶电泳分析以及染料法所需要的试剂等,都要经过测定和校准[20-21]。引物的优化、引物浓度、退火温度都会影响到PCR的效率,进而影响试验的质量。
通过标准曲线的建立,来评价PCR的扩增效率。定量标准物可以是cDNA、质粒DNA、特定生物体样本的RNA或DNA以及是世界上公认的生物学标准物。对于定量标准的稀释MIQE也做了详细的阐述,另外归一化是可信赖的RT-qPCR的重要组成部分[22-23]。目前,常用△△Cq法对样品和Ref⁃erence RNA基因不同浓度进行归一化,从而可以控制由抽提率、反转率和扩增效率产生的差异[24]。但是目的基因和对照基因必须在相似的扩增效率下。MIQE规定,在所提交的文章中,不仅要有标准曲线,还要有注明斜率值,扩增效率=10-1/slope-1。
扩增效率的高低可以直接出PCR问题,低的扩增效率(<90%)可能是由于Tag酶的活性低,抑制剂的污染,退火温度未优化,引物设计不合理或扩增子含二级结构;过高扩增效率(>105%)的原因一般是由于引物二聚体或非特异性扩增,不过人为的操作不当移液器的不准或不当操作、离心不当等而影响扩增效率的高低是经常发生的。
在绘制标准准曲线中,样品稀释倍数应确保覆盖试验中可能用到的模板浓度,宽的动态范围是必须的。理想状态下,扩增曲线的分布是均一的,间隔也代表着每次扩增的倍数,扩增效率应保持在90%~105%。标准曲线的r或R2可代表符合回归曲线的程度,RT-qPCR理想的r绝对值应>0.990或R2>0.998。使用Excel电子表格添加趋势线功能,以确定每对引物的使用cDNA动态浓度。
6 数据分析
数据分析方法因所选定量方法的差异而有所差异。绝对定量是通过样品的Cq值与标准曲线进行比较得到的;相对定量是一定量的试验组和对照组中目的基因的相对比率。无论是绝对定量还是相对定量,试验数据的校准是必不可少的;绝对定量中每次的试验标准样品必须与待测样品同时平行扩增;而相对定量在比较多个样本时,选择一个样品作为对照样本,其他所有样本目标基因的表达都以对照样本上调或下调,一般以基准或未处理样本作为对照。
6.1 内参基因
在RT-qPCR试验中,内参基因被用来作为数据标准化的对照,以校正作为模板得cDNA所存在得数量差异[25]。一个好的内参基因应确保在不同的组织、不同的处理条件下表达都无变化。往往在试验中,要选用多个内参基因进行比较,而选择好的内参基因,才能使试验数据被信服。不同的条件或时间点下△Cq≤0.5。建议在试验中同时检测至少3个或3个以上位于不同代谢途径中的特定内参基因,并对表达差异最小的几个基因进行几何平均以准确标准化数据[26-29]。
内参基因确定以后,可以采用Livak法,也就是2-△△Ct法、参照基因的△Ct法或Pfaffl法进行分析,每种方法都有优缺点。
6.2 试验重复性与重现性
试验的重复性与重现性是必须考虑的因素,因为在试验中有两种可变因素可能会影响到试验的准确性和重复性。分别是生物学差异和技术差异。生物学差异是不同个体或组织样品间基因表达的差异所致;技术差异是试验药品、器材不同和操作不当等人为因素导致。为了减少生物学和技术差异对试验带来的影响,可以通过试验设计加以避免。一般除了设置对照组,每个样本还要设置3个生物学重复,并且每个重复要设置至少两个技术重复,另外,再加上两个无模板(NTC)对照。如果试验是在对照组和处理组之间进行的比较,设置的3个生物学重复样品应该来源于独立试验中分别进行处理的样品[30-31]。
7 展 望
实时荧光PCR技术自建立以来,发展迅速、应用广泛,表现出许多优势。近些年来,许多科研工作者基于荧光PCR的基本原理对荧光PCR技术进行不断深入的研究和改进,使荧光PCR技术得到了进一步的完善,并在此基础上开发出了许多新的荧光PCR技术。随着标准化的实时荧光PCR技术的不断发展该技术将在生物学、医学基础研究以及试验动物学等广泛的领域中得到更加广泛的推广应用。
[1] Bustin S A,Benes V,Garson J A,et al.The MIQE guidelines:min⁃imum information for publication of quantitative real-time PCR experiments[J].Clin Chem,2009,55(4):611-622.
[2] Taylor C F,Paton N W,Lilley K S,et al.The minimum informa⁃tion about a proteomics experiment(MIAPE)[J].Nat Biotechnol,2008,25(8):887-893.
[3] Field D,Garrity G,Gray T,et al.The minimum information about a genome sequence(MIGS)specification[J].Nat Biotechnol,2007,26(5):541-547.
[4] Echeverri C J,Beachy P A,Baum B,et al.Minimizing the risk of reporting false positives in large-scale RNAiscreens[J].Nat Meth⁃ods,2006,3(10):777-779.
[5] Haney S A.Increasing the robustness and validity of RNAiscreens[J].Pharmacogenomic,2007,8(8):1 037-1 049.
[6] Sansone S A,Fan T,Goodacre R,et al.The metabolomics stan⁃dards initiative[J].Nat Biotechnol,2007,25(8):846-848.
[7]Brazma A,Hingamp P,Quackenbush J,et al.Minimum informa⁃tion about a microarray experiment(MIAME)-toward standards for microarray data[J].Nat Genet,2001,29(4):365-371.
[8] Taylor C F,Field D,Sansone S A,et al.Promoting coherent mini⁃mum reporting guidelines for biological and biomedical investiga⁃tions:the MIBBI project[J].Nat Biotechnol,2008,26(8):808-889.
[9] Burns MJ,Valdivia H,Harris N.Analysis and interpretation of da⁃ta from real-time PCR trace detection methods using quantitation of GM soya as a model system[J].Analytical and Bioanalytial Chemistry,2004,378(6):1 616-1 623.
[10]Burns M J,Nixon G J,Foy C A,et al.Standardisation of data from real-time quantitative PCR methods-evaluation of outliers and comparisonof calibration curves[J].BMC Biotechnol,2005(5):31.
[11] Ellison S L,English C A,Burns M J,et al.Routes to improving the reliability of low level DNA analysis using real-time PCR[J].BMC Biotechnol,2006(6):33.
[12] Burns M J,Valdivia H.Modelling the limit of detection in re⁃al-time quantitative PCR[J].Eur Food Res Technol,2007,226(6):1 513-1 524.
[13] Lefever S,Hellemans J,Pattyn F,et al.RDML:Structured lan⁃guage and reporting guidelines for real-time quantitative PCR da⁃ta[J].Nucleic Acid Res,2009,37(7):2 065-2 069.
[14] Fleige S,Pfaffl M W.RNA integrity and the effect on the realtime qRT-PCR performance[J].Mol Aspects Med,2006,27:126-139.
[15] Imbeaud S,Grandens E,Bonlaneper V,et al.Towards standardiza⁃tion of RNA quality assessment using userindependent classifiers of microcapillary electrophoresis traces[J].Nucleic Acid Res,2005,33-56.
[16] Denisov V,Strong W,Walder M,et al.Development and valida⁃tion of RQI:An RNA puality indicator for the Experion automated electrophoresis system[J].Bio-Rad Bulletin,2008,1 108:1 103-1 108.
[17]Zuker M.MFOLD web server for nucleic acid folding and hybridiza⁃tion prediction[J].Nucleic Acid Res;2003,31(13):3 406-3 415.
[18] Pattyn F,Robbrecht P,De Paepe A,et al.RTPrimerDB:the re⁃al-time PCRprimer and probe database,major update[J].Nucleic Acids Res,2006,34(1):684-688.
[19] Pattyn F,Speleman F,De Paepe A,et al.RTPrimerDB:the re⁃al-time PCR primer andprobe database[J].Nucleic Acids Res,2003,31(1):122-123.
[20] Nolan T,Hands R E,Bustin S A.Quantification of mRNA using real-time RT-PCR[J].Nat Protoc,2006(1):1 559-1 582.
[21] Ririe K M,Rasmussen R P,Wittwer C T.Product differentiation by analysis of DNA melting curves during the polymerase chain reaction[J].Anal Biochem,1997,245(2):154-160.
[22]Cronin M,Ghosh K,Sistare F,et al.Universal RNA reference ma⁃terials for gene expression[J].Clin Chem,2004,50(8):1 464-1 471.
[23] Novoradovskaya N,Whitfield M L,Basehore L S,et al.Universal Reference RNA as a standard for microarray experiments[J].BMC Genomics,2004(5):20.
[24] Hellemans J,Mortier G,De Paepe A,et al.qBase relative quantifi⁃cation framework and software for management and automated analysis of real-time quantitative PCR data[J].Genome Biol,2007(8):19.
[25] Gutierrez L,manriat M,Guemin S,et al.The lack of a systematic validation of reference genes:a serious pitfall undervalued in re⁃verse transcription-ploymerase chain reaction(RT-PCR)analy⁃sis in plants[J].Plant Biotechnol,2008,6(6):609-618.
[26] Vandesompele J,et al.Accurate normalization of real-time quan⁃titative RT-PCR data by geometric averaging of multiple inter⁃nal control genes[J].Genome Biol,2002,34:1-11.
[27] Expósito-Rodríguez M,Borges A A,Borges-Pérez A,et al.Selec⁃tion of internal control genes for quantitative real-time RT-PCR studies during tomato development process[J].BMC Plant Biol,2008,22(8):131.
[28] Artico S,Nardeli S M,Brilhante O,et al.Identification and evalua⁃tion of new reference genes in Gossypium hirsutum for accurate normalization of real-time quantitative RT-PCR data[J].BMC Plant Biol,2010,21(10):49.
[29] Barsalobres-Cavallari C F,Severino F E,Maluf M P,et al.Identi⁃fication of suitable internal control genes for expression studies in Coffea arabica under different experimental conditions[J].BMC Mol Biol,2009,6(10):1.
[30] Willems E,Leyns L,Vandesompele J.Standardization of realtime PCR gene expression data from independent biological repli⁃cates[J].Anal Biochem,2008,379:127-129.
[31] Vandesompele J D P K,Pattyn F,Poppe B,et al.Accurate nor⁃malization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes[J].Genome Biol,2002(3):7.
