不同人群的SNPs基因型数据中r2值对TagSNP数量的影响*
2012-03-11张镏琢郑娟娟孙远洁袁重胜
张镏琢 秦 平 李 昂 郑娟娟 孙远洁 袁重胜 刘 艳△
研究人员通过对比病例组和对照组的基因型频率的不同来发现基因对疾病的影响,由于单体型(haplotype)整合了各个多态位点之间的连锁不平衡(linkage disequilibrium,LD)信息,因此基于单体型的关联研究比基于单个单核苷酸多态性(single nucleotide polymorphisms,SNPs)的分析能够提供更高的检出功效〔1-2〕。与疾病相关的基因型很有可能就存在于病例组和对照组之间基因型频率不同的染色体区域内,理论上对全部的SNPs位点都进行基因分型,就能够寻找到这样的区域〔3〕,但应用这种方法进行鉴定的成本却过于昂贵。已有研究表明〔4〕,用少部分的遗传标记仍可保留单体型的大部分信息,例如单体型图计划已鉴定出约50万个标签SNP(TagSNP)位点,提供了与一千万个SNPs位点大致相同的图谱信息,从而大幅度地减少了成本使得研究易于进行。目前,已有几种根据基因分型或单体型数据筛选tagSNP的方法〔5〕,研究者利用SNPs及其他遗传变异在染色体上的组成特点,从HapMap中获得tagSNP的信息,通过软件分析用来定位与重要医学特征相关的基因〔6〕。TagSNP的筛选大大简化了基因与疾病的关联分析,使遗传疾病的基因定位能够更为有效的进行。
材料与方法
1.数据来源
利用国际人类基因组单体型图计划(The International HapMap Project,http://www.hapmap.org) 的通用基因组浏览器(HapMap Genome Btowser)下载2009年2月公布的Ⅲ期22号染色体的SNPs基因型数据。其中,北京汉族人群(Han Chinese in Beijing,China,CHB)包括84个样本的18 588个SNPs信息;东京日本人群(Japanese in Tokyo,Japan,JPT)包括86个样本的18 103个SNPs信息;而来自欧洲北部和西部的犹他州人群(Utah residents with Northern and Western European ancestry from the CEPH collection,CEU)则包括165个样本的19 660个SNPs信息。
2.软件介绍
在SNPs基因型数据分析中,常用的软件有BEST、GeneDigger、HaploBlock 以及 Haploview,其中 Haploview是由剑桥大学编写的利用已有的基因型数据来计算连锁不平衡统计量并推断人群单体型模型的公用软件包,目前的最新版本为Haploview4.2(可从http://www.broadinstitute.org/haploview/haploview-downloads免费下载),可在1.4版及以上的Java运行环境(JRE)中使用。
Haploview中的Tagger计算机程序提供了筛选tagSNP的方法〔7〕,主要参数是次等位基因频率(Minor Allele Frequency,MAF)阈值以及 tagSNP和未检测SNP之间的最小连锁不平衡相关程度(用r2值来表示),即侯选基因上所有常见 SNPs(即次等位基因频率大于阈值者),要么作为tag-SNP进行直接检测,要么与一个待检SNP相关联(r2值超过设定值)。此外,研究者也可以结合现有文献选取感兴趣的可能的功能位点(如基因编码区和调控区的多态位点)作为tagSNP。
3.筛选tagSNP
运行Haploview4.2分别读取HapMap中的CHB、JPT和CEU的22号染色体SNPs基因型数据,之后首先要对数据的信息进行整理,设定Hardy-Weinberg平衡切断值(HW p-value cutoff)为0.0010,次等位基因频率(minimum minor allele freq)的最小值为0.0010,未缺失的基因型频率最小值为75%,最大孟德尔遗传规律错误的个数(max mendel errors)为1,Haploview将最终选取满足这些条件的SNPs进行有效的分析;随后在tagSNP的筛选过程中设定不同的r2值(0.5~1.0),并记录tagSNP的数量。
4.TagSNP数量的比例关系
HapMap所提供的不同人群的相同染色体的SNPs数量并不完全相同,为了排除所筛选的tagSNP数量会受到人群总SNPs数量的影响,需要计算不同人群中不同r2值所对应的tagNP数量占总SNPs数量的百分比例。
结 果
运行Haploview4.2分别处理三个人群的SNPs基因型数据,不同r2值所对应的tagSNP数量及其占总SNPs数量的百分比例的具体情况见表1。当r2值为0.8(软件默认值)时,在CHB中筛选出7 752个tag-SNP,这表示由占总SNPs数量41.70%的这7 752个tagSNP所构建的单体型即可反映北京汉族人群整个22号染色体上的18 588个SNPs的大部分信息;与此相比,依然是在 r2值为0.8时,分别需要7 460个(41.21%)和9086个(46.22%)tagSNP才能反映东京日本人群和来自欧洲北部和西部的犹他州人群22号染色体上的单体型信息。即使在相邻的SNPs位点存在着完全连锁的情况下(r2值为1)所筛选的tagSNP的数量还是明显的低于总SNPs数量,此时CHB、JPT和CEU的tagSNP数量分别占总SNPs数量的65.56%、63.35%和70.05%。
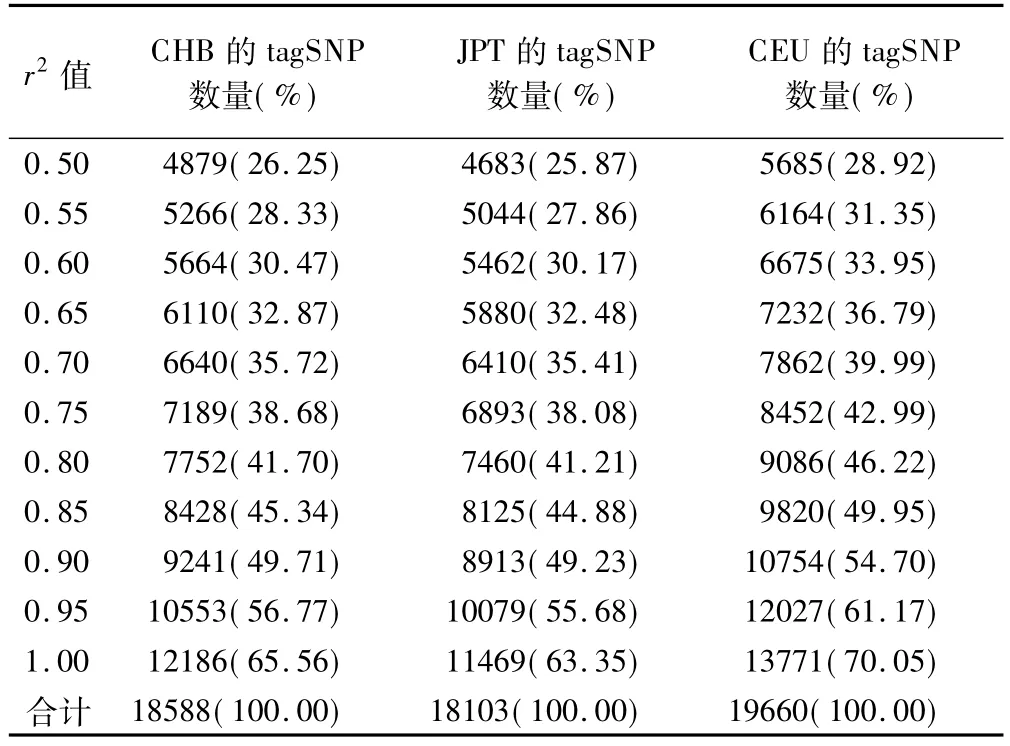
表1 三个人群中不同r2值所对应的tagSNP数量及其占总SNPs数量的百分比例
若以设定的r2值为横坐标,以所筛选的tagSNP数量或其占总SNPs数量百分比例为纵坐标做图,则可以更为直观地看到三个人群中北京汉族人群和东京日本人群比较接近,且三个人群均表现出随着所设定的r2值的增大tagSNP数量及其百分比例呈现出逐渐增多的趋势;但在r2值相同时三个人群的tagSNP数量却存在着一定的差异,表现为日本人群的数量最少,北京汉族人群其次,欧洲北部和西部的犹他州人群的数量最多。在三个人群中tagSNP数量的增加速度都表现为在r2值小于0.9时较为平稳,r2值大于0.9时增加速度都有所提高,其中以北京汉族人群的增加更为明显。
讨 论
运用Haploview软件进行tagSNP的筛选,可直接分析从HapMap上下载到的阶段性SNPs基因型数据,通过计算连锁不平衡统计量来推断人群单体型模型。有研究显示利用HapMap基因型数据所筛选的tagSNP进行关联研究比单位点研究的统计效力更高,且可不受数据库完整性的影响〔7〕。本研究即是运用Haploview4.2软件计算出三个人群(北京汉族人群、东京日本人群和来自欧洲北部和西部的犹他州人群)22号染色体在不同r2值(最小连锁不平衡相关程度)时所对应的tagSNP数量及其占总SNPs数量的百分比例,结果显现出tagSNP的数量受r2值的影响较大,虽然三个人群在SNPs连锁不平衡关系上存在着一定的差异(相对说来,北京汉族人群和东京日本人群的结果较为接近),但均表现出随着所设定的r2值的增大tagSNP数量及其百分比例呈现出逐渐增多的趋势,不过tagSNP数量的增加速度在三个人群中略有不同。究其增加速度不同的原因,一方面在于HapMap计划所检测的三个人群总SNPs位点个数以及样本例数的差异(北京汉族人群和东京日本人群样本例数几乎相等,而欧洲北部和西部的犹他州人群的样本例数则几乎是北京汉族人群和东京日本人群样本例数的总和),理论上较大的样本量意味着位点间有更多的重组机会(重组机会的增多可降低原位点间的r2值),故当固定r2值以筛选tagSNP时,较大的样本量就可导致有更多的tagSNP被筛选出来;另一方面,影响r2值的因素还包括群体有效大小(Ne)以及位点间重组率(c),三个人群不同的遗传背景使得位点间重组率和重组率图(相邻的SNPs位点间的重组率即组成了重组率图) 产生差异,因 E(r2)=1/(1+4Nec)〔8〕,从而也导致了三个人群的tagSNP在数量及增加速度上的差异。所以,在实际研究中要依据目标人群、研究经费等方面的具体情况设置合理的r2值,若因研究经费有限需要检测的SNPs位点数量较少时,可通过降低r2值来减少tagSNP的数量,尤其是当研究人群不同时,不应盲目地遵从由参考样本数据所计算出的r2值,应更相信从实际研究数据中所计算得到的r2值。此外,当估算出位点间的r2值后,还可进一步推断群体有效大小,因为群体有效大小是一个重要参数,有助于解释人群是如何进行演变和扩大的,并可提高对复杂性状遗传模式的理解和建模〔9〕。
目前,在疾病的病因学研究中,尽管已在发现疾病的危险等位基因方面取得了很大的进步,但大多数的遗传风险仍然无法解释。在广泛的人群中全面的检测所有常见和稀有的等位基因可对疾病的遗传风险有一个更为完整的认识〔8〕。但对全部SNPs位点进行关联研究确实存在着诸多困难(工作量大、成本较高等),而利用少量的tagSNP就能提供与全基因组SNPs位点大致相同的图谱信息的方式,则可以良好的解决这些问题。相信,随着生命科学的不断发展和进步,提取tagSNP的算法将会得到逐步完善,从而在分子流行病学和生命科学的研究中发挥积极的作用。
1.Akey J,Jin L,Xiong M.Haplotypes vs single marker linkage disequilibrium tests:what do we gain?Eur J Hum Genet,2001,9(4):291-300.
2.邹莉玲,赵耐青,秦国友,等.应用关联规则筛选疾病相关的SNP位点及其组合的分析方法.中国卫生统计,2009,26(3):226-233.
3.Johnson GC,Esposito L,Barratt BJ,et al.Haplotype tagging for the identification of common disease genes.Nat Genet,2001,29(2):233-237.
4.Patil N,Berno AJ,Hinds DA,et al.Blocks of limited haplotype diversity revealed by high-resolution scanning of human chromosome 21.Science,2001,294(5547):1669-1670.
5.Gopalakrishnan S,Qin ZS.TagSNPselection based on pairwise LD criteria and power analysis in association studies.Pac Symp Biocomput,2006,511-522.
6.Xu Z,Kaplan NL,Taylor JA.Tag SNPselection for candidate gene association studies using HapMap and gene resequencing data.Eur J Hum Genet,2007,15(10):1063-1070.
7.Bakker PI,Yelensky R,Pe'er I,et al.Efficiency and power in genetic association studies.Nat Genet,2005,37(11):1217-1223.
8.Sved JA.Correlation measures for linkage disequilibrium within and between populations.Genet Res(Camb),2009,91(3):183-192.
9.Tenesa A,Navarro P,Hayes BJ,et al.Recent human effective population size estimated from linkage disequilibrium.Genome Res,2007,17(4):520-526.
