伤寒沙门菌核糖核酸酶G对胞内非编码RNA T3956水平的影响
2012-01-26王菲孟彦辰詹莉芳张晓磊张海方生秀梅徐顺高黄新祥
王菲,孟彦辰,詹莉芳,张晓磊,张海方,生秀梅,徐顺高,黄新祥
近年来,随着生物信息学和分子生物学技术的不断发展,细菌中的非编码RNA(non-coding RNA,ncRNA)作为一类新发现的基因表达调控因子,已受到越来越多的关注[1]。过去长期认为RNA仅是把遗传信息从DNA带到蛋白质的一个过渡产物,然而目前大量的研究陆续发现细菌中一些非编码小RNA,作为应答环境压力的调节元件[2],在细菌的物质代谢、环境适应、群体感应和细菌毒力等方面发挥着重要的调节作用[3-6]。绝大多数非编码小RNA通过与靶mRNA配对,在转录后水平影响靶基因mRNA的翻译或(和)稳定性,从而调节目的基因的表达,影响细胞的多种生理功能[2,7]。在细菌体内,这些ncRNA分子的活力主要受控于其胞内的水平。对大肠埃希菌和其他一些菌属的研究表明,核糖核酸酶 E、G、Ⅲ主要参与对 ncRNA水平的调控[8]。因此,研究ncRNA分子在菌体内的水平是更好了解ncRNA作用的前提。
伤寒沙门菌(Salmonella enterica serovar Typhi,S.Typhi)是一种严重的人类肠道致病菌,也是一种重要的研究原核基因表达与调控的模式生物。本室通过对伤寒沙门菌野生株GIFU10007在普通LB、氧应激、高酸、高盐等不同生长条件下的基因转录谱分析、测序、生物信息学预测和实时定量PCR(qRTPCR)验证,从中挑选出一个位于基因ftsX和t3957之间的非编码区ncRNA分子,命名为T3956。我们通过实时定量PCR分析初步发现,在伤寒沙门菌RNase E、RNase G、RNaseⅢ三种主要核糖核酸酶的缺陷变异株中,T3956的胞内水平在核糖核酸酶G基因(rng)缺陷株中变化较为明显。因此,本研究拟通过构建伤寒沙门菌rng缺陷变异株和rng缺陷回补株,并利用qRT-PCR分析RNase G对伤寒沙门菌胞内ncRNA T3956水平的影响。
1 材料与方法
1.1 材料
1.1.1 菌株和质粒 野生型S.Typhi GIFU10007、E.coli SY372λpir和自杀质粒pGMB151由日本岐阜大学医学院微生物学教研室馈赠;E.coli DH5α由本室保存;pGEM-T载体为Promega公司产品。
1.1.2 主要试剂与材料 DNA聚合酶 ExTaq、rTaq、限制性内切酶 BamH I、Xho I、BglⅡ、T4DNA连接酶、dNTP、DNase I、实时 PCR 均为 TaKaRa(大连)公司产品;DNA聚合酶pfu、RNase G为Fermentas(上海)公司产品,X-Gal、L-阿拉伯糖、氨苄西林为Sigma(美国)公司产品,TA克隆试剂盒为Promega(北京)公司产品;质粒提取试剂盒、胶回收试剂盒为Axygen(美国)公司产品;总RNA提取试剂盒为Qiagen(德国)公司产品;逆转录试剂盒为Invitrogen(美国)公司产品。
1.1.3 主要仪器 凝胶成像分析系统(Syngene);PCR扩增仪2720 Thermal Cycler(ABI);核酸检测仪Speetrophotometer ND-100(NanoDrop);电转化仪Gene Pulsero II(BIO-RAD9);超速冷冻离心机Centifuge 5417R(Eppendorf);荧光定量PCR仪CFX96TMReal-Time System(Bio-Rad)。
1.2 方法
1.2.1 引物设计 根据本室测定的伤寒沙门菌野生株GIFU10007基因组序列信息(未公布),利用Oligo6软件设计rng缺陷株引物、rng缺陷回补株引物和t3956 qRT PCR引物。rng缺陷株引物设计:依据GiFU10007基因序列显示,rng基因全长1 488 bp,在rng基因上游和下游设计两对特异性PCR引物F1A/F1B和F2A/F2B,以S.Typhi GIFU10007为模板,扩增出340 bp和728 bp的同源性片段(分别用F1,F2表示)。在F1B、F2A的5'端共有20个碱基互补配对通过PCR用以定向连接F1和F2片段,在F1A、F2B的5'端加BamH I酶切位点用以克隆F1和F2连接片段至自杀质粒。rng缺陷回补株引物设计:在rng基因上下游设计引物FA,FB,并在5'端分别加上 Xho I及 BglⅡ酶切位点用以连接pBAD/gⅢ载体。t3956 qRT PCR引物设计:根据本室对伤寒沙门菌野生株GIFU10007基因组中的t3956基因测序信息,在序列中间设计一段特异性引物,通过检测荧光值来观察该基因的表达情况。所用引物均由上海生工生物技术服务有限公司合成,序列见表1。

表1 引物序列Tab 1 Sequences of primers
1.2.2 伤寒沙门菌rng缺陷株的制备 伤寒沙门菌rng缺陷变异株的制备过程主要参考文献[9]。以伤寒沙门菌野生株基因组DNA为模板,用特异性引物F1A/F1B和F2A/F2B分别扩增出rng基因上下游同源性片段F1和F2。由于在F1B、F2A的5'端共有20个碱基互补配对,用酚仿-乙醇法纯化后的F1和F2片段共同作为模板通过PCR获得F1-F2定向连接产物。胶回收F1-F2片段后与pGMET载体连接,热击法导入E.coli DH5α,先用BamH I酶切和PCR检测对疑似阳性克隆质粒作初步鉴定,再用DNA序列分析(测序由上海生工生物技术服务有限公司完成)予以进一步鉴定。用BamH I酶切阳性重组pMD18-T质粒,胶回收酶切片段(F1-F2),通过T4DNA连接酶将其连接至自杀质粒pGMB151的BamH I酶切位点,用热击法导入E.coli SY372λpir,筛选疑似阳性克隆并用BamH I酶切和PCR检测鉴定。通过试剂盒提取带有目的片段F1-F2的阳性自杀质粒,用电击法导入伤寒沙门菌野生株,并在5%蔗糖LB平板上进行同源重组。用引物F1A、F2B扩增同源性片段观察重组现象,将连续4次传代完全重组的菌株作为S.Typhi rng缺陷株。
1.2.3 伤寒沙门菌rng基因缺陷回补株的制备根据伤寒沙门菌野生株基因组序列信息,在rng基因上下游设计引物FA、FB,并在5'端分别加上Xho I及BglⅡ酶切位点,用高保真的DNA聚合酶pfu扩增出目的片段。纯化目的片段后用Xho I及BglⅡ对其进行双酶切反应,通过T4DNA连接酶将其与经同样酶切的pBAD/gⅢ质粒4℃过夜连接。用热击法将酶切产物转化至E.coli DH5α,筛选阳性克隆并用酶切和PCR进行初步鉴定,再用DNA测序分析验证(测序由上海英骏生物技术服务有限公司完成)。试剂盒提取测序正确的阳性克隆质粒和空质粒pBAD/gⅢ(阴性对照),分别电击导入伤寒沙门菌rng缺陷变异株中,命名为rng缺陷回补株 Δrng(pBAD-rng),空质粒对照株 Δrng(pBAD)。
1.2.4 细菌培养及总 RNA提取 分别挑取S.Typhi野生株、rng缺陷株、rng缺陷回补株和空质粒株单菌落于1 ml等渗LB培养液中,37℃振荡(250 r/min)培养过夜,然后以1∶100分别转接于20 ml等渗LB培养液中,37℃振荡(250 r/min)培养至 D(600 nm)值各为0.2,0.8 和1.2,其中培养回补株和回补空质粒株的LB液体中需加入L-阿拉伯糖和氨苄西林,终浓度分别为0.5 mg/ml和100 μg/ml。将培养管冰上放置 10 min后离心(4 000 r/min,10 min,4℃)收集菌体,再用TE缓冲液洗菌体1次,离心(4 000 r/min,10 min,4℃)收集菌体。用总RNA提取试剂盒分别提取细菌总RNA,并用无RNA酶的DNA酶 I(37℃ 30 min,80℃ 2 min)消化残余DNA,用核酸检测仪检测RNA,确定其浓度,同时进行琼脂糖凝胶电泳,分析RNA的质量(要求rRNA条带清晰且无残余DNA)。
1.2.5 RNA逆转录与实时定量PCR(qRT-PCR)采用上述方法提取细菌总RNA后用特异性引物逆转录成 cDNA。逆转录体系和条件:4 μg总 RNA,4 μl特异性引物 t3965FB,1 μl dNTP 和 4 μl无 RNA 酶的ddH2O混合后65℃作用5 min,冰浴1 min,再加入 5 × Fs 缓冲液 4 μl,0.1 mol/L DTT 1 μl,RNase OUT 1 μl,SuperScript Ⅲ 1 μl,总体积 20 μl,逆转录反应条件为25℃10 min,50℃50 min,70℃15 min。然后按照TaKaRa公司的实时定量PCR试剂盒操作说明进行实验。qRT-PCR的反应体系:逆转录产物 1 μl,t3956 上下游引物各 1 μl,混合物(含有 ExTaq DNA聚合酶、dNTP、PCR缓冲液和SYBR Green)17 μl。实验重复3次,每次设置平行对照组,得到的qRT-PCR结果用One-way ANOVA进行统计分析。
2 结果
2.1 成功制备伤寒沙门菌rng缺陷变异株
以S.Typhi野生株GIFU10007的基因组DNA为模板,用特异性引物F1A/F1B和F2A/F2B进行PCR扩增,获得了rng基因上、下游同源性片段F1和F2,分别为340 bp和728 bp(图1a)。通过PCR得到长度约1 068 bp的F1-F2定向连接片段后进行TA克隆,对筛选得到的阳性克隆经酶切和PCR鉴定后,再进行DNA测序分析,结果显示F1-F2连接产物序列与S.Typhi野生株GIFU10007的rng基因序列一致。将F1-F2片段成功克隆至自杀质粒后(图1b),提取带有目的片段的自杀质粒电击转入S.Typhi野生株GIFU10007,然后在5%蔗糖LB平板上进行同源重组,得到仅有缺失972 bp的小片段菌株后,在普通LB平板上连续4次传代均只有小片段出现(图1c),表明S.Typhi rng缺陷变异株制备成功。
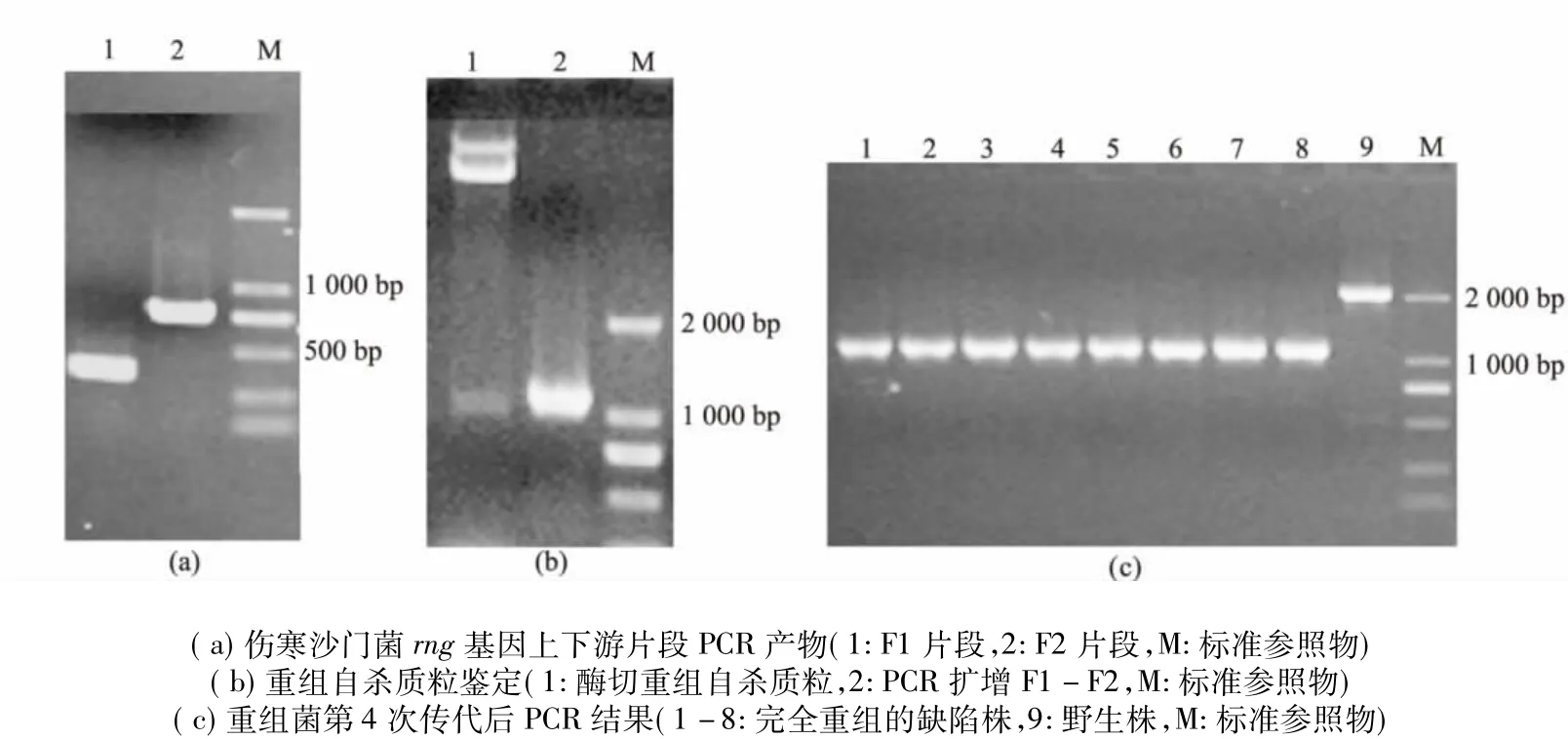
图1 伤寒沙门菌rng基因缺陷变异株的制备Fig 1 Preparation of the rng deleted mutant of S.Typhi
2.2 成功制备伤寒沙门菌rng缺陷回补株
以野生株基因组DNA为模板,用引物FA/FB进行PCR扩增,获得rng基因回补片段大约1 588 bp(图2a)。将扩增的目的片段经双酶切后与表达载体pBAD/gⅢ进行连接,热击导入 E.coli DH5α中,对疑似阳性克隆进行酶切和 PCR鉴定后(图2b),再经DNA测序分析验证,测序结果显示插入的回补片段与目的基因完全一致。试剂盒提取测序正确的阳性克隆质粒和空质粒pBAD/gⅢ(阴性对照),分别电击导入伤寒沙门菌rng缺陷变异株中,用目的基因特异性引物FA/FB对回补株进行PCR扩增,结果显示在回补株中获得了目的基因的特异片段(图2c),说明含rng基因的载体成功转入缺陷株中,即S.Typhi rng缺陷回补株制备成功。
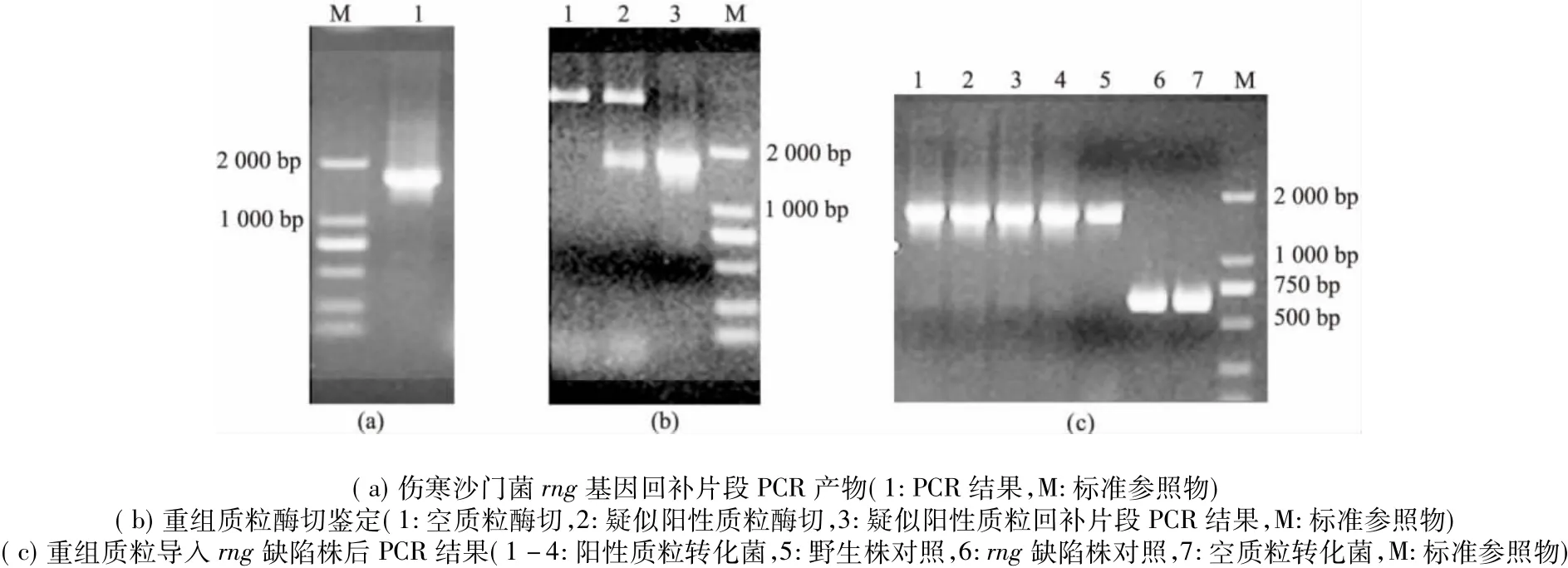
图2 伤寒沙门菌rng基因缺陷回补株的制备Fig 2 Rescue of rng in the rng deleted mutant of S.Typhi
2.3 qRT-PCR分析RNase G对胞内T3956水平的影响
为探讨RNase G对伤寒沙门菌ncRNA T3956表达的影响,我们分别提取伤寒沙门菌D(600 nm)值为0.2,0.8 和1.2 时的总 RNA,并用 qRT-PCR 分析各时相野生株、rng缺陷株、回补株和空质粒株中T3956的水平。结果(图3)显示,当细菌处于对数早期即D(600 nm)为0.2时,各菌株的胞内T3956水平并无显著差异;当D(600 nm)为0.8及1.2即对数生长中后期和稳态期时,rng缺陷株的T3956胞内水平均较野生株明显增加(P<0.01)。在各时相中,回补株与野生株之间、空质粒菌株与缺陷株之间的T3956胞内水平相似。结果表明,在伤寒沙门菌中RNase G可影响胞内ncRNA T3956的水平,并且在细菌对数生长中后期和稳态期时RNase G对T3956水平的影响更为明显。
3 讨论
核糖核酸酶类对ncRNA的成熟、降解和胞内水平起着重要的调控作用。目前,在大肠埃希菌中已发现的核糖核酸酶类有20多种[10]。在细菌中主要的核糖核酸酶有RNase E、RNase G和RNaseⅢ。RNase E是主要作用于单链RNA的核糖核酸内切酶,其羧基末端还可以与其他的酶类连接组装成降解复合体,参与许多小RNA的降解[11]。RNase G由rng基因编码,与RNase E起催化作用的氨基末端具有同源性。同RNase E一样,RNase G主要降解单链RNA,并且多水解5'末端含有单磷酸基团的RNA和富含AU碱基的区域[12]。在大肠埃希菌中,RNase G和RNase E共同参与16S rRNA 5'端的成熟加工。尽管RNase G与RNase E的氨基末端具有同源性,但是两者作用的酶切位点并不完全一致[13]。RNaseⅢ是一种作用于双链RNA的核糖核酸内切酶,对sRNA和靶mRNA配对的双链区进行切割,可以同时降解sRNA和其配对的靶mRNA[14]。另外,参与小RNA调控的核糖核酸外切酶还有聚核甘酸磷酸化酶(PolynucleotidePhosphorylase,PNPase)和聚腺苷酸聚合酶(PolyA polymerase I,PAP I)。PNPase和RNase E一起参与组成降解体,参与小RNA降解[15]。PAP I的多聚腺苷化作用可以影响大肠埃希菌GlmY sRNA的稳定性[16]。

图3 qRT-PCR分析伤寒沙门菌核糖核酸酶G对非编码RNA的影响Fig 3 qRT-PCR was performed to analyze the influence of RNaseG on the cellular level of non-coding RNA T3956 in S.Typhi.
在原核生物中,非编码小RNA的胞内水平和生理作用往往受到多种核糖核酸酶的共同调控,不同核糖核酸酶的作用方式也各不相同[17]。例如,对鼠伤寒沙门菌的一种非编码小RNA MicA的研究表明,其胞内周转调控涉及两种不同的作用机制,当MicA与靶mRNA配对后,RNaseⅢ可以同时降解MicA sRNA和靶mRNA,而对于未配对的MicA sRNA来说,RNase E则是参与游离MicA降解的主要因子,同时RNase E还可以招募其他因子如PNPase来参与游离MicA的周转,控制MicA sRNA的胞内水平[18]。这些核糖核酸酶共同参与调控MicA sRNA转录后的水平,使其达到最适的水平,从而发挥对靶mRNA的调节作用。
本文研究了伤寒沙门菌RNase G对胞内ncRNA T3956水平的影响,实时定量PCR结果表明RNase G可以影响胞内ncRNA T3956的水平,并且在细菌的对数生长中后期和稳态期作用更加明显,提示伤寒沙门菌中的RNase G对ncRNA T3956的胞内水平和周转更新发挥着重要的调节作用,这对研究ncRNA T3956的生理功能有着重要的意义。但RNase G对T3956发挥降解作用的具体酶切位点和作用机制尚不清楚,以及有无其它的核糖核酸酶共同参与对胞内ncRNA水平的调控,还有待进一步的研究。
[1]Vogel J,Sharma CM.How to find small non-coding RNAs in bacteria[J].Biol Chem,2005,386(12):1219-1238.
[2]Storz G,Opdyke JA,Zhang A.Controlling mRNA stability and translation with small non-coding RNAs[J].Curr Opin Microbiol,2004,7(2):140-144.
[3]Grke B,Vogel J.Noncoding RNA control of the making and breaking of sugars[J].Genes Dev,2008,22(21):2914-2925.
[4]Mass E,Gottesman S.A small RNA regulates the expression of genes involved in iron metabolism in Escherichia coli[J].Proc Natl Acad Sci USA,2002,99(7):4620-4625.
[5]Lenz DH,Mok KC,Lilley BN,et al.The small RNA chaperone Hfq and multiple small RNAs control quorum sensing in Vibrio harvevi and Vibrio cholerae[J].Cell,2004,118(1):69-82.
[6]Romby P,Vandenesch F,Wagner EG.The role of RNAs in the regulation of virulence-gene expression[J].Curr Opin Microbiol,2006,9(2):229-236.
[7]Guillier M,Gottesman S.Remodelling of the Escherichia coli outer membrane by two small regulatory RNAs[J].Mol Microbiol,2006,59(1):231-247.
[8]Viegas SC,Pfeiffer V,Sittka A,et al.Characterization of the role of ribonucleases in salmonella small RNA decay[J].Nucleic Acids Res,2007,35(22):7651-7664.
[9]茅凌翔,朱超望,黄新祥,等.伤寒沙门菌phoP基因缺陷变异株的制备[J].江苏大学学报:医学版,2007,17(2):145-149.
[10]Arraiano CM,Andrade JM,Dominques S,et al.The critical role of RNA processing and degradation in the con-trol of gene expression[J].FEMS Microbiol Rev,2010,34(5):883-923.
[11]Carpousis,AJ,Van Houwe G,Ehretsmann C,et al.Copuri cation of E.coli RNAase E and PNPase:evidence for a specific association between two enzymes important in RNA processing and degradation[J].Cell,1994,76(5):889-900.
[12]Jiang X,Belasco JG.Catalytic activation of multimeric RNase E and RNase G by 5'-monophosphorylated RNA[J].Proc Natl Acad Sci USA,2004,101(25):9211-9216.
[13]Tock MR,Walsh AP,Carroll G,et al.The CafA protein required for the 5'-maturation of 16S rRNA is a 5'-end-dependent ribonuclease that has context-dependent broad sequence specificity[J].J Biol Chem,2000,275(12):8726-8732.
[14]Afonyushkin T,Vecerek B,Moll I,et al.Both RNase E and RNase III control the stability of sodB mRNA upon translational inhibition by the small regulatory RNA RyhB[J].Nucleic Acids Res,2005,33(5):1678-1689.
[15]Andrade JM,Arraiano CM.PNPase is a key player in the regulation of small RNAs that control the expression of outer membrane proteins[J].RNA,2008,14(3):543-551.
[16]Reichenbach B,Maes A,Kalamorz F,et al.The small RNA GlmY acts upstream of the sRNA GlmZ in the activation of glmS expression and is subject to regulation by polyadenylation in Escherichia coli[J].Nucleic Acids Res,2008,36(8):2570-2580.
[17]Viegas SC,Arraiano CM.Regulating the regulators:How ribonucleases dictate the rules in the control of small noncoding RNAs[J].RNA Biol,2008,5(4):230-243.
[18]Viegas SC,Silva IJ,Saramago M,et al.Regulation of the small regulatory RNA MicA by ribonuclease III:a targetdependent pathway[J].Nucleic Acids Res,2011,39(7):2918-2930.
