智力发育障碍的研究进展和启示
2012-01-08姜永辉,周文浩,罗飞宏
罗飞宏教授 很高兴美国杜克大学医学院儿科遗传及神经科姜永辉教授来复旦大学附属儿科医院讲学,受《中国循证儿科杂志》编辑部委托,邀请姜永辉教授就“智力发育障碍(intellectual developmental disorder, IDD)”组织一期专家对谈录,也十分高兴地邀请到对这一话题很感兴趣的复旦大学附属儿科医院新生儿科周文浩教授。
1 MR的定义及流行病学资料
罗飞宏教授 IDD过去也称精神发育迟缓(mental retardation,MR),MR其最初判定标准仅为IQ<70,目前将其内涵拓展为成年之前出现的两种或以上适应性行为的缺陷和显著的认知功能受损。姜教授能介绍一下为什么将原来MR重新定义为IDD。
姜永辉教授 MR是美国精神病学会(APA)制定的《美国精神疾病诊断标准》DSM-Ⅳ及之前的版本中所用的医学术语。DSM-Ⅴ开始广泛使用智力障碍(intellectual disability,ID)这个医学名词。美国智力落后协会(The American Association on Intellectual and Development Disability,AAIDD)将ID定义为一种功能障碍,以便与WHO制定的《国际功能、障碍与健康分类》(ICF)保持一致,而在DSM-Ⅴ中使用的智力发育障碍(intellectual developmental disorder,IDD)则与国际疾病分类法(ICD-11)相一致,DSM-Ⅴ将IDD定义为始于发育过程中的认知能力的缺陷,包括当前存在的智力障碍和发育过程中出现的适应能力的障碍,须满足以下3个标准:①智力发育障碍以一般的心智能力的缺陷为特征,如推理、解决问题、规划、抽象思维、判断、理论和经验学习等;②个人年龄和社会文化背景的适应能力减弱,指一个人在以下一个或多个方面的日常生活中,如沟通、社会参与、个人独立性、社会交往、学校或工作中的作用、家庭或社区中始终需要支持;③所有的症状必须始于发育阶段。
罗飞宏教授 根据DSM-Ⅳ的分类,85%为轻度精神发育落后(IQ 50~70),该类患者可以获得相当于六年级水平的学术技能;约10%为中度精神发育落后(IQ 35~55);3%~4%为重度精神发育落后(IQ 20~40)。其患病率因各调查所规定的定义、诊断标准、取样方法和心理学检验方法的不同而有差异。
姜永辉教授 根据AAIDD和WHO的报道,儿童IDD患病率为1%~2%。按照这个比例,中国IDD患儿可多达1 300多万,对家庭和社会来说均是沉重的负担。
2 IDD的病因
罗飞宏教授 IDD的病因复杂多样,包括生物医学因素和社会心理文化原因。前者指脑在发育过程中(产前和围生前期)接受到的各种不利因素,可使脑的发育不能达到应有水平,最终影响智力。后者指文化剥夺、教养不当和感觉剥夺等因素可使后天信息输入不足或不适当,从而影响智力水平。
周文浩教授 以往临床工作中遇到IDD患儿,通常会归咎于新生儿期的过失,如宫内窘迫、产伤、产时窒息和生后高危因素暴露等。目前已有数据显示, 约30%的IDD可能与遗传因素有关,而70%则与父母、围生期因素和环境等非遗传因素有关。
姜永辉教授 根据常规遗传性疾病的分类,绝大多数遗传病为单基因病和染色体病,其中绝大多数的遗传代谢病也属于单基因病的范畴。在过去的5~10年随着基因芯片技术在临床医学领域的广泛运用,使IDD的遗传病因检测成功率显著提高,而之前的近50年只能借助染色体核型分析技术对染色体数目及结构异常性疾病进行检测,检测片段在6 Mb以上,基因芯片技术则可将检测片段缩小至数十甚至数个kb。随着分子诊断技术的不断发展,从基因的点突变(point mutation),到全外显子测序(whole exome sequencing),再到全基因组测序(whole genome sequencing),新的病因在不断被发现,如2012年在NEngJMed发表的1篇文章中又新报道了12个与IDD相关的基因拷贝数变异(表1)。
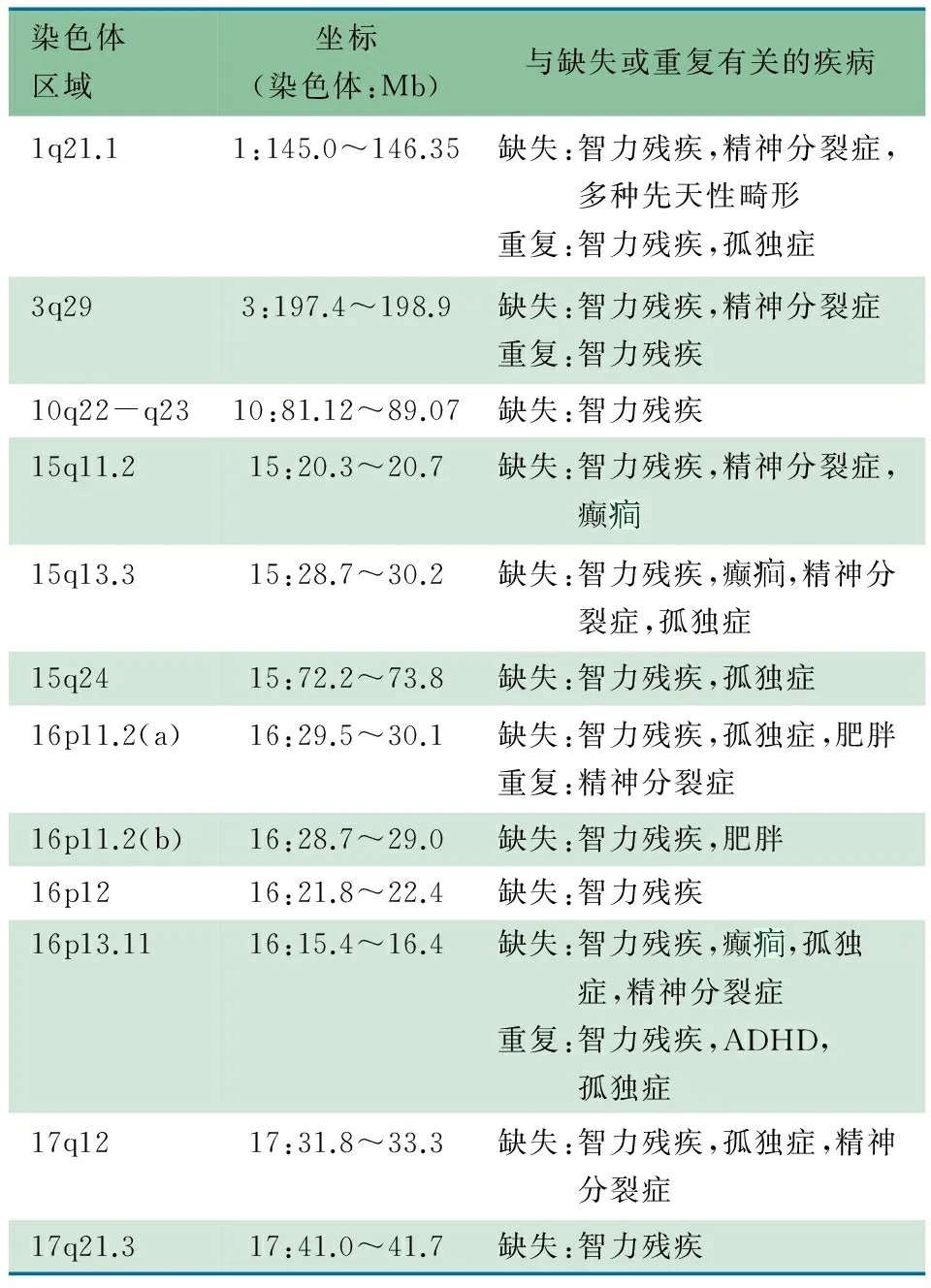
表1 目前报道的与IDD相关的拷贝数变异
在女孩IDD的遗传因素中以Rett综合征最常见,男孩则以Fragile X综合征最常见,占IDD患儿的21%以上染色体核型分析+基因组芯片杂交技术+基因突变分析等分子诊断技术的综合运用,使IDD遗传因素的检出率上升至50%~70%,但仍有相当一部分IDD患儿是由于非遗传因素(如产伤等)所致,而对于轻度IDD患儿的非遗传因素,如环境因素等通常难以发现。
罗飞宏教授 目前有资料显示,在引起IDD的众多遗传病因中,3类疾病高居榜首,分别是唐氏综合征、Fragile X综合征和胎儿酒精综合征(fetal alcohol syndrome,FAS)。
周文浩教授 FAS是母亲在妊娠期间酗酒对胎儿造成的永久出生缺陷,其最主要的影响是永久的中枢神经系统损害。西方国家发病率达1/3 000,在中国目前没有由于母孕期烟草、酒精和毒物等异常胎儿暴露因素所导致的宫内发育迟缓精确的发病率数据。
3 IDD的病理生理学研究
罗飞宏教授 IDD的病因复杂多样,借助目前的分子诊断技术可找出部分与遗传相关的病因,但不同病因导致的结局存在病理机制上的相似性。最近有文献报道,微小RNA与神经发育存在密切的关系,该研究发现了4种可调控视网膜祖细胞向视网膜上不同神经元分化的微小RNA(miR-129, miR-155, miR-214, miR-222)。也有文献报道在脑细胞分化的研究中,微小RNA的Dicer酶敲除后,可引起相应的浦肯野细胞的凋亡和坏死。如何去理解这种脑发育机制上的病理生理改变?
姜永辉教授 导致IDD的病理生理机制目前仍不明确,动物模型研究发现与神经元突触的可塑性及突触数目有一定的关系,但在人体内研究存在相当的难度,一是动物(如小鼠等模型)与人的智力相差甚远,二是可获得的人体脑发育研究标本有限。近年来对脑发育研究运用较多的是诱导多能干细胞(induced pluripotent stem cells,iPS),比如将动物或者人的皮肤细胞诱导成神经元细胞,从而在细胞水平研究与临床表现相关的病理生理机制,研究进展速度很快但仅局限于细胞水平,而人体的智力调控是一个复制的网络。
罗飞宏教授 在脑的众多功能中,学习和记忆非常关键,这些方面的研究有何最新进展?
姜永辉教授 对于学习和记忆方面的研究,主要集中在突触水平,包括突触的发育和调控等。但脑的功能是一个复杂的调控网络,并非局限于单个细胞或单个通路,也涉及到行为、饮食、学习和记忆等诸多方面的功能,对于这些功能调控的脑机制研究有助于找到相应的治疗干预措施,但目前尚无准确的答案。
周文浩教授 动物实验确实与人体研究存在很大差距,在动物实验中许多的实验条件(特殊分泌因子、营养因子和功能训练等)都是可控制的,可以观察到脑局部区域的突触重建等变化,但很难在人体直观地观察到。
姜永辉教授 借助先进的影像诊断技术如PET、弥散张量成像(DTI)及功能MRI,可比较不同区域脑组织的功能活动,但都是描述性的发现,得出的只是差别,而不能给出其病理生理的变化机制。
4 IDD的遗传学诊断和治疗探索
周文浩教授 目前在临床工作中,最容易接触到IDD患儿的应该是神经科、儿童保健科和发育儿科,如果这些专科医生的遗传学知识背景薄弱,对于遗传因素导致的IDD就很难识别出来,早期发现遗传信息的机会被延迟。国外是否有一个可供借鉴的诊断流程可以将IDD这一大类疾病有效地聚焦到遗传因素上?
姜永辉教授 在美国,因IDD而到遗传科或发育行为科就诊的患儿,都要经历比较规范的流程性诊断(图1),比较基因组芯片杂交技术(array CGH)是基本的遗传病因筛选技术。通常会有初级保健儿科医生(primary care pediatrician,PCP)对患儿进行智能发育的评估,一方面PCP可以直接给患儿开出某些遗传检测项目,比如染色体核型分析等,有异常发现后再进一步转诊给遗传专科医生;另一方面对于严重IDD患儿,特别是一些伴有特殊面容或多发畸形的患儿,则通常直接转诊给遗传专科医生。
中国目前较欠缺的是儿科医生对儿童不同发育阶段评估的培训和遗传知识的普及,具备相应的遗传学知识会使其在今后的临床工作中能对这些疾病遗传信息进行早期识别和做出遗传学诊断。
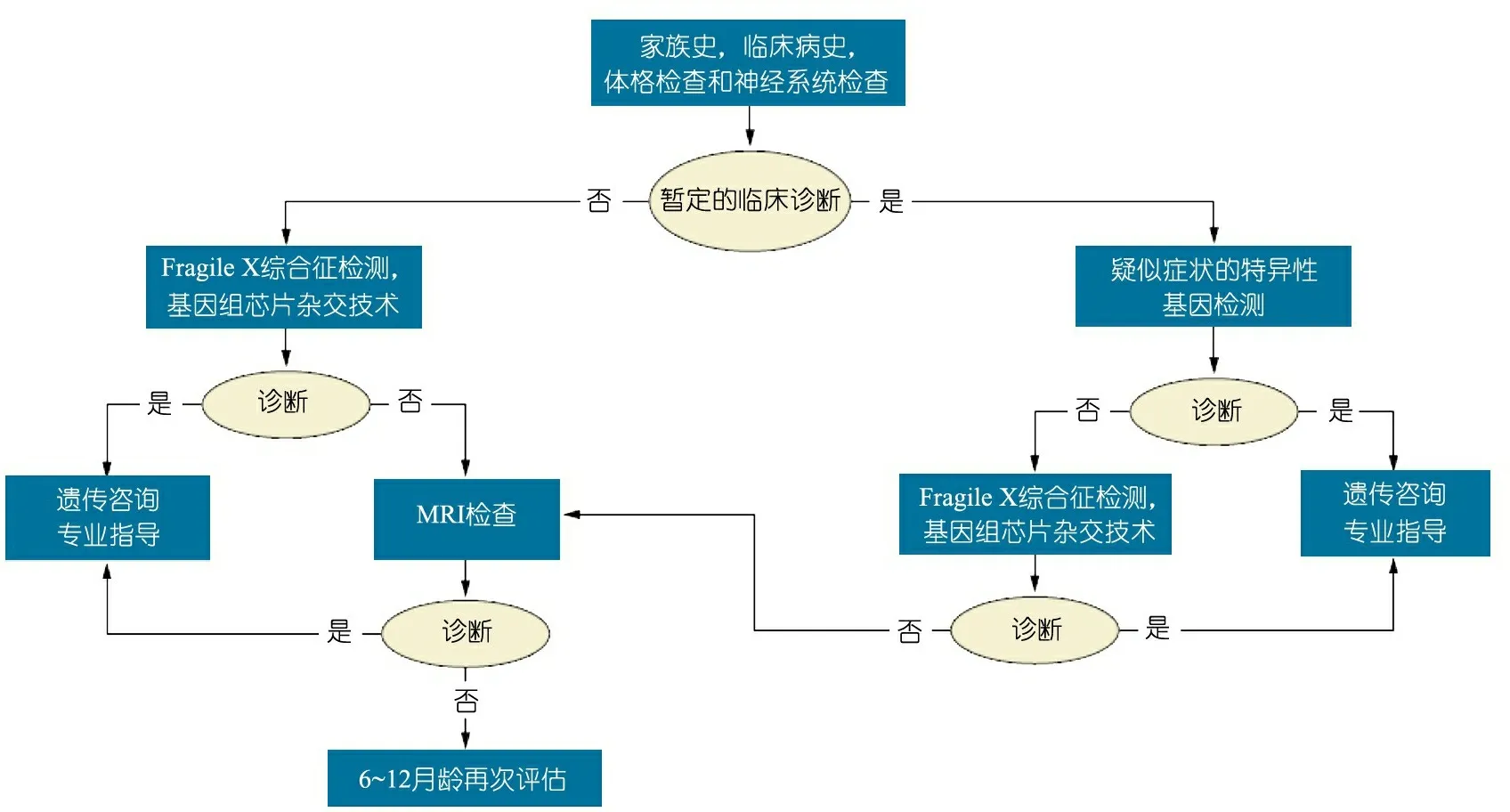
图1 原因不明的IDD患儿的评估和诊断筛选流程
周文浩教授 随着分子诊断技术的迅猛发展,如新一代测序技术(next-generation sequencing,NGS)等,在不久的将来是否会有针对某些导致IDD的遗传性疾病的筛查“套餐”可供临床使用?
姜永辉教授 这是一种发展趋势,国外目前已经有一些商业化的筛查“套餐”供科研和临床使用,价格也在可承受范围内。
周文浩教授 对于某些遗传代谢病,通过新生儿期症状前的筛查,可避免或减轻IDD的程度,但代谢病在IDD患儿中仅占很小一部分,大部分为单基因病或微缺失/微重复(microdeletion/microduplication)畸形患儿,只有当临床表型出现后才可能被识别。儿科医生最感兴趣的还是希望借助先进的检测手段进行早期筛查。苯丙酮尿症(PKU)作为新生儿期筛查的典型疾病,主要归因于低苯丙氨酸奶方的研制成功,也即有效治疗手段的获得。对于糖原累积病、黏多糖病等溶酶体贮积病,是否也有在新生儿期即给予筛查并早期干预的可能?
姜永辉教授 现在美国法定的29种新生儿期筛查的疾病,理论上绝大多数都有干预的手段,干预效果可能存在差异,有的疾病可得到完全控制或改善,而有的则是部分改善,要将溶酶体贮积病等纳入新生儿期筛查,最关键的还是要有可干预的办法。对于那些发生率较高且可致严重IDD的遗传性疾病,明确病因并给予适当的遗传咨询是关键。在对Rett综合征致病基因的动物实验研究中已发现,当正常基因被输送回动物体内后,完全逆转了该疾病模型的致死表现型,但动物模型是否能完全模拟人体的自然病程,以及体内基因治疗方面的研究仍面临巨大挑战。如果能找到IDD的病因及相应的有效干预手段,结合上述分子诊断技术,才是真正意义上实现了对IDD的早期筛查和干预。
罗飞宏教授 从传统观念上看,IDD后天性的干预措施几乎缺乏,但新近的一些动物研究发现似乎带来了曙光。在果蝇模型中,2-甲基-6-苯基乙炔嘧啶可有效防止细胞突触缺失和行为的学习和记忆障碍。IDD志愿者给予氯苯咪脲(Fenobam)单剂量口服50~150 mg,可以观察到患者的感觉门控、注意力和抑制力得到某种程度的改善。2011年罗氏公司针对18~30岁的唐氏综合征患者开展了一种新型药物的Ⅰ期临床试验,观察药物对唐氏综合征患者学习、记忆、语言和安全性的影响。一些新的给药载体如通过转基因给药方式、干细胞等也在研究中,姜教授你的看法如何?
姜永辉教授 基因治疗多数还处于Ⅰ期临床试验阶段,面临的最重要的问题包括:基因如何导入?能否穿透血脑屏障改善脑发育或功能?目的基因导入的靶向位点在哪?还有包括导入基因的剂量问题。
周文浩教授 目前是否有一种遗传性疾病的研究模式,包括遗传病因的探索、发病机制的研究、干预手段的发现,可作为某一类导致IDD的遗传病的临床识别和治疗的借鉴?

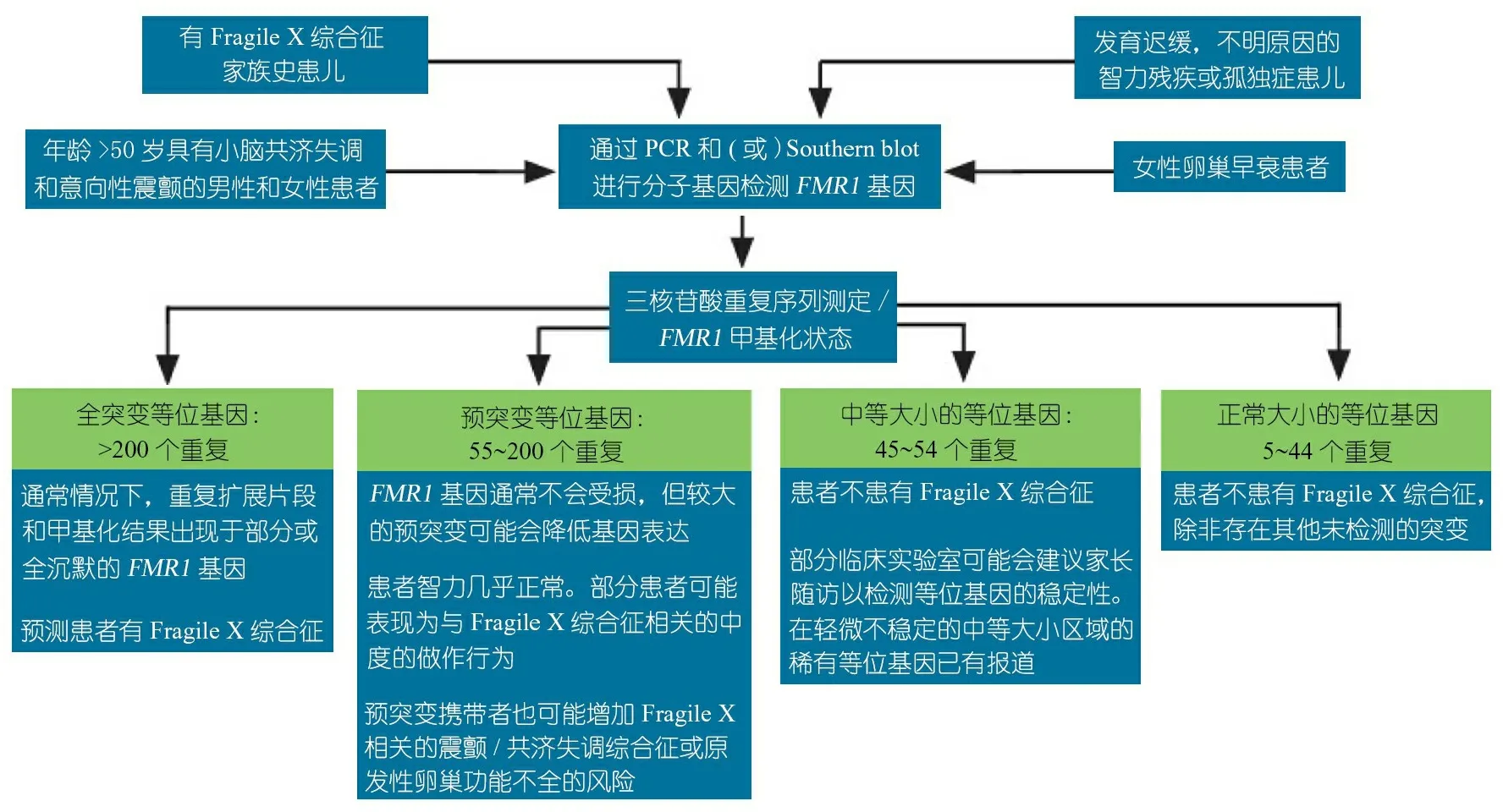
图2 与FMR1基因相关的疾病检测筛选流程
当FMR1基因发生突变时会阻碍其编码蛋白FMRP表达,导致大脑中FMRP缺失。几年前,美国麻省理工学院的神经学教授Mark Bear发现在正常情况下FMRP蛋白可以控制或阻断大脑细胞中mGluR5激活的信号途径;FMRP缺失时,代谢性谷氨酸受体5(mGluR5)信号过度激活,促发过量突触蛋白合成,从而导致大脑神经元联系异常以及与Fragile X综合征相关的行为及认知障碍。进一步的Fragile X综合征小鼠动物模型研究显示,mGluR5拮抗剂可逆转树突棘异常、惊厥发作和认知行为问题,从而显示出良好的治疗前景。因此对IDD遗传病因的寻找,并在此基础上开展发病机制的研究,目的是为了找到有效干预的办法。
罗飞宏教授 很荣幸邀请到2位教授就IDD的遗传病因、早期识别的重要性、现有的分子诊断技术和规范的诊断流程进行深入的交流,从分享Fragile X综合征的研究历程中启示如下:①IDD病因复杂,遗传因素占有相当大的比率;②借助分子诊断技术可早期进行识别;③遗传机制的研究将有助于治疗手段的探寻和发现。再次感谢两位教授与中国的儿科医生分享IDD的研究历程。
致谢 感谢复旦大学附属儿科医院陆炜博士承担本文的录音整理工作。
